Welcome to the Plant Breeding and Genomics Webinar Series
|
|
|
- Job McBride
- 10 years ago
- Views:
Transcription
1 Welcome to the Plant Breeding and Genomics Webinar Series Today s Presenter: Dr. Candice Hansey Presentation: Host: Heather Merk Technical Production: John McQueen PBG home page: Sign up for PBG News:

2 Please fill out the survey evaluation! (You will be contacted via ) Watch past webinars and sign up for future webinars!

3 How to Align Sequences Presenter: Candice Hansey Michigan State University

4 Overview NavigaAng NCBI to obtain sequences Using BLAST for sequence alignment Using other programs for specialized sequence alignment Next generaaon sequence alignment programs

5 Overview NavigaAng NCBI to obtain sequences Using BLAST for sequence alignment Using other programs for specialized sequence alignment Next generaaon sequence alignment programs

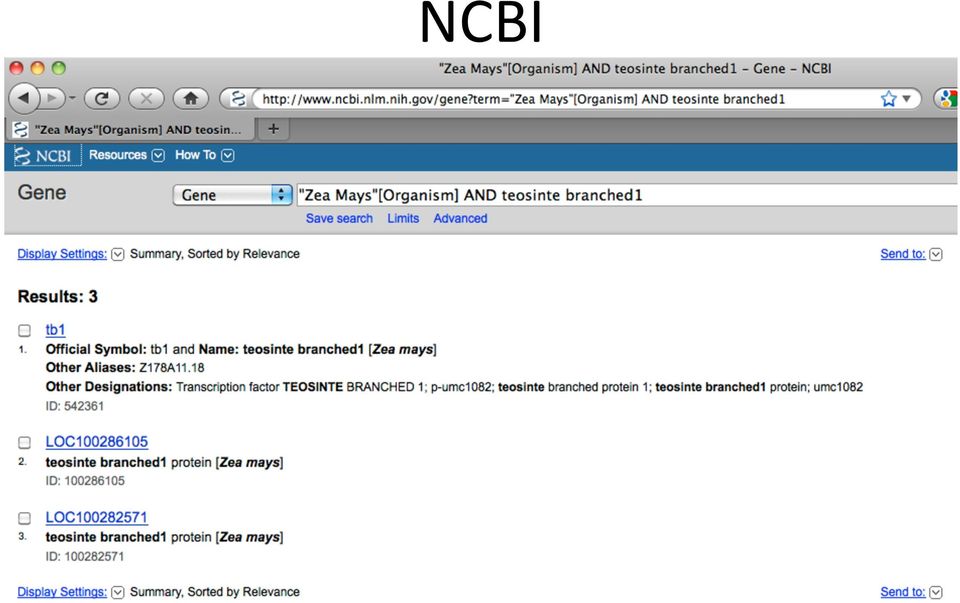
6 Goal Obtain sequence for the maize teosinte branched1 gene and then find organisms with orthologous genes

7 NCBI
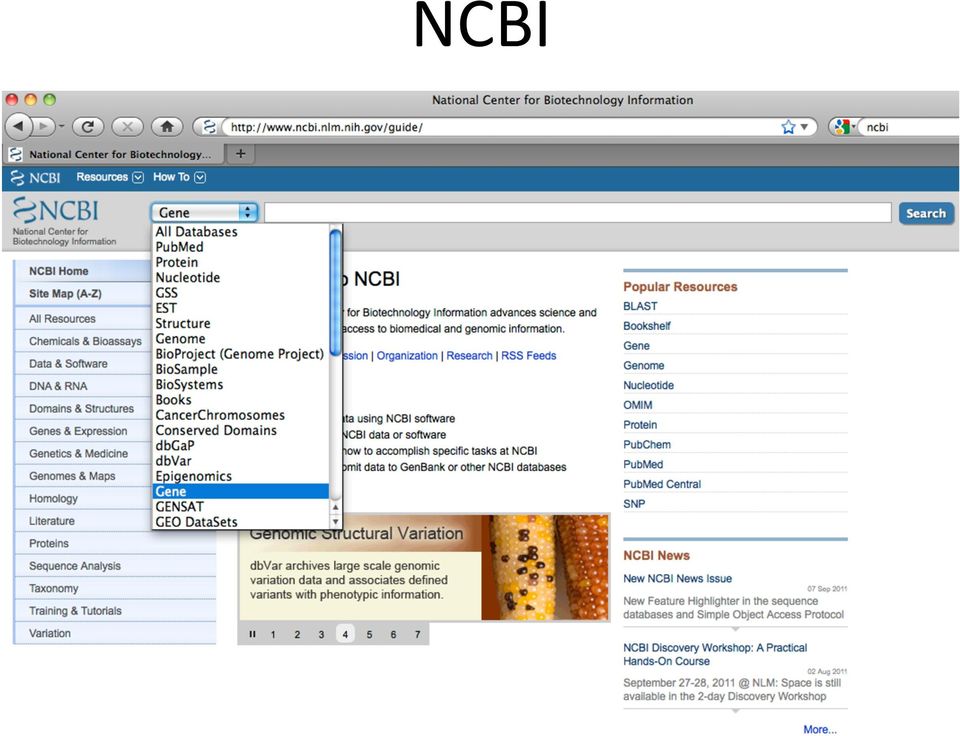
8 NCBI Search specifying Zea Mays as the organism resulted in thousands of sequences

9 NCBI
10 NCBI

11 NCBI
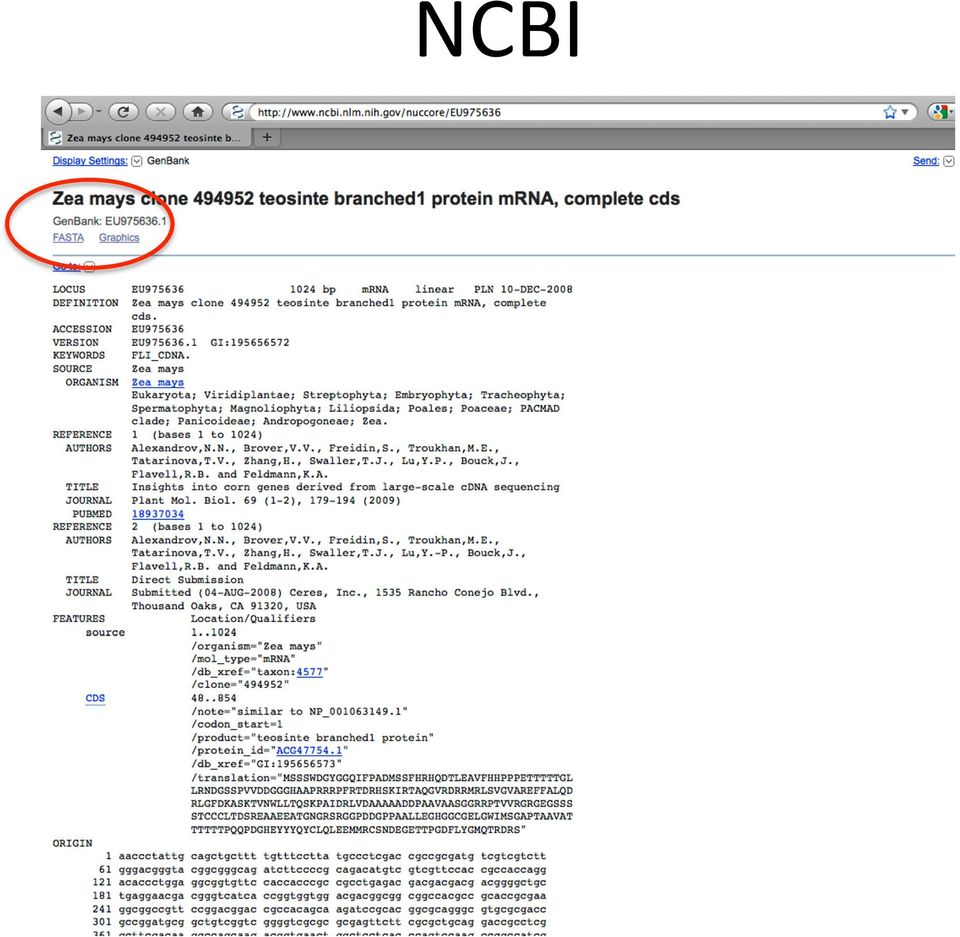
12 NCBI

13 Overview NavigaAng NCBI to obtain sequences Using BLAST for sequence alignment Using other programs for specialized sequence alignment Next generaaon sequence alignment programs

14 BLAST BLAST = Basic Local Alignment Search Tool

15 BLAST BLAST = Basic Local Alignment Search Tool Used for searching a query sequence or sequences against a database or subject sequence

16 BLAST BLAST = Basic Local Alignment Search Tool Used for searching a query sequence or sequences against a database or subject sequence Uses a local alignment, which searches for regions in the query that locally align to subject sequences

17 BLAST BLAST = Basic Local Alignment Search Tool Used for searching a query sequence or sequences against a database or subject sequence Uses a local alignment, which searches for regions in the query that locally align to subject sequences BLAST uses word based heurisacs to approximate the Smith- Waterman algorithm to find the near- opamal local alignments quickly, thus gaining speed over sensiavity.

18 BLAST BLAST = Basic Local Alignment Search Tool Used for searching a query sequence or sequences against a database or subject sequence Uses a local alignment, which searches for regions in the query that locally align to subject sequences BLAST uses word based heurisacs to approximate the Smith- Waterman algorithm to find the near- opamal local alignments quickly, thus gaining speed over sensiavity. Two main versions: NCBI Blast and WU- BLAST

19 Local vs Global Alignments For sequences that are divergent, the opamal global alignment introduces gaps that can hide biologically relevant informaaon such as moafs.

20 BLAST The process starts by searching the sequences for exact matches of small fixed length strings from the query called words.

21 BLAST The process starts by searching the sequences for exact matches of small fixed length strings from the query called words. These matches are the seeds for the local alignments.
22 BLAST The process starts by searching the sequences for exact matches of small fixed length strings from the query called words. These matches are the seeds for the local alignments. The next step is to extend the seed by aligning the sequence unal a gap is found mismatches are inserted here.
23 BLAST The process starts by searching the sequences for exact matches of small fixed length strings from the query called words. These matches are the seeds for the local alignments. The next step is to extend the seed by aligning the sequence unal a gap is found mismatches are inserted here. A gapped alignment is then performed using a modified Smith- Waterman algorithm indels are added here.
24 BLAST The process starts by searching the sequences for exact matches of small fixed length strings from the query called words. These matches are the seeds for the local alignments. The next step is to extend the seed by aligning the sequence unal a gap is found mismatches are inserted here. A gapped alignment is then performed using a modified Smith- Waterman algorithm indels are added here. Only results scoring above a threshold (expect value or e value) are reported back to the user
25 BLAST Programs blastn - query DNA, subject DNA blastp- query Protein, subject Protein blastx - query DNA (6 frame translaaon) subject Protein tblastn - query protein - subject DNA (6 frame translaaon) - slow tblastx - query DNA (6 frame translaaon), subject DNA (6 frame translaaon) - very slow
26 Which Program Should You Use What database do you have and how sensiave does your search has to be? blastn, blastp good for idenafying sequences that are already in a database, finding local regions of similarity in closely related organisms.
27 Which Program Should You Use What database do you have and how sensiave does your search has to be? blastn, blastp good for idenafying sequences that are already in a database, finding local regions of similarity in closely related organisms. blastx - Use when you have a nucleoade sequence with an unknown reading frame and/or sequencing errors that would lead to frame shi`s or coding errors such as ESTs. More sensiave.
28 Which Program Should You Use What database do you have and how sensiave does your search has to be? blastn, blastp good for idenafying sequences that are already in a database, finding local regions of similarity in closely related organisms. blastx - Use when you have a nucleoade sequence with an unknown reading frame and/or sequencing errors that would lead to frame shi`s or coding errors such as ESTs. More sensiave. tblastn- useful for finding homologs in sequence where the frame is unknown or sequencing errors are likely to be present such as ESTs and dra` sequence.
29 Which Program Should You Use What database do you have and how sensiave does your search has to be? blastn, blastp good for idenafying sequences that are already in a database, finding local regions of similarity in closely related organisms. blastx - Use when you have a nucleoade sequence with an unknown reading frame and/or sequencing errors that would lead to frame shi`s or coding errors such as ESTs. More sensiave. tblastn- useful for finding homologs in sequence where the frame is unknown or sequencing errors are likely to be present such as ESTs and dra` sequence. tblastx - used to detect novel ORFs/exons. Very Slow, use as the last resort.
30 Performing BLAST
31 Performing BLAST
32 Performing BLAST
33 Output from BLAST
34 Output from BLAST
35 Output from BLAST
36 BLAST For addiaonal informaaon on BLAST go to hdp:// blast.ncbi.nlm.nih.gov/blast.cgi?cmd=web&page_type=blastdocs
37 BLAST For addiaonal informaaon on BLAST go to hdp:// blast.ncbi.nlm.nih.gov/blast.cgi?cmd=web&page_type=blastdocs Web based BLAST interfaces are designed for low throughput searching Personalized databases can be generated and subsequent BLASTs performed using the command line Using the command line BLAST, mulaple sequences can be aligned simultaneously
38 Ubuntu For PC users I highly recommend gegng Ubuntu This can be run as a virtual machine on your PC through programs such as VirtualBox hdps://
39 Overview NavigaAng NCBI to obtain sequences Using BLAST for sequence alignment Using other programs for specialized sequence alignment Next generaaon sequence alignment programs
40 EST and cdna Alignment Exonerate is a generic tool for pairwise sequence comparison (hdp:// and comes grid ready with the ability to chunk files directly through exonerate
41 EST and cdna Alignment Exonerate is a generic tool for pairwise sequence comparison (hdp:// and comes grid ready with the ability to chunk files directly through exonerate Exonerate can be run with many different models for gapped and ungapped alignments
42 EST and cdna Alignment Exonerate is a generic tool for pairwise sequence comparison (hdp:// and comes grid ready with the ability to chunk files directly through exonerate Exonerate can be run with many different models for gapped and ungapped alignments The est2genom model can be used for alignment of EST sequences to genomic sequence and the cdna2genome model can be used to align full length cdnas that can be flanked by UTRs
43 EST and cdna Alignment Exonerate is a generic tool for pairwise sequence comparison (hdp:// and comes grid ready with the ability to chunk files directly through exonerate Exonerate can be run with many different models for gapped and ungapped alignments The est2genom model can be used for alignment of EST sequences to genomic sequence and the cdna2genome model can be used to align full length cdnas that can be flanked by UTRs For addiaonal models see the man page online at hdp:// ~guy/exonerate/exonerate.man.html or on the command line with the - h opaon exonerate - h
44 EST and cdna Alignment Command line example exonerate - - query est_sequences.fasta - - querytype DNA - - target genome_sequence.fasta targedype DNA - - model est2genome showalignment no - - showvulgar no - - showtargetgff no - - ryo %qi\t%a\t%qab\t%qae\t%tab\t%tae \n minintron maxintron 1500 >exonerate_alignment.txt
45 EST and cdna Alignment Command line example exonerate - - query est_sequences.fasta - - querytype DNA - - target genome_sequence.fasta targedype DNA - - model est2genome showalignment no - - showvulgar no - - showtargetgff no - - ryo %qi\t%a\t%qab\t%qae\t%tab\t%tae \n minintron maxintron 1500 >exonerate_alignment.txt The output from this command will be a tab delimited file with the following columns for each alignment: query_id, target_id, query_start, query_end, target_start, target_end Note: using this output format the posiaons are in interbase coordinates For complete list of opaons see the man page online at hdp:// exonerate/exonerate.man.html or on the command line with the - h opaon
46 Whole Genome Alignment MUMmer designed for rapid alignment of enare genomes NUCmer designed for alignment of conags to another set of conags or a genome PROmer designed for alignment of species too divergent for DNA alignment using six- frame translaaon of both input sequences hdp://mummer.sourceforge.net/
47 Whole Genome Alignment mummer can handle mulaple reference and query sequences Command line usage mummer [opaons] <reference- file> <query- files> mummerplot [opaons] <match file> Example command line mummer mum b c genotype1.fasta genotype2.fasta > output.mums - mum finds all maximal unique matches, - b will compute forward and reverse complement matches, - c reports all match posiaons relaave to the forward starnd mummerplot x [0,275287] y [0,265111] png output.mums - x sets the x- axis range in the plot, - y sets the y- axis range in the plot, - - png outputs the plot in.png format AddiAonal opaons are available in the manual or by using the - h opaon for both mummer and mummerplot Examples for NUCmer and PROmer are available at hdp://mummer.sourceforge.net/ examples/
48 Whole Genome Alignment Forward MUMs are red and reverse MUMs are green. Dots on a line with a slope = 1 are unchanged, and dots on a line with a slope = - 1 are inverted. hdp://mummer.sourceforge.net/examples/
49 Short Sequence Alignment Vmatch is a tool that is ideal for aligning short sequences such as probes, primers, and SNPs with short context sequence This program requires a license hdp://
50 Overview NavigaAng NCBI to obtain sequences Using BLAST for sequence alignment Using other programs for specialized sequence alignment Next generaaon sequence alignment programs
51 High Throughput Sequencing Plaxorms Illumina HiSeq 1000 and HiSeq 2000 Illumina Genome Analyzer IIx Life Sciences/Roche 454 pyrosequencing Pacific Biosciences Ion Torrent
52 High Throughput Sequencing HiSeq 2000 Highly parallel sequencing by synthesis Single and paired- end reads between 50 bp and 100 bp 187 million single end or 374 million paired- end reads per lane High error rate in the 3 end
53 Sequence File Format Read Name Sequence Quality Quality scores are in ASCII characters that are converted to Phred scores. These scores provide a likelihood that the base was called incorrectly in 10 chance the base call is incorrect 20 1 in 100 chance the base call is incorrect 30 1 in 1000 chance the base call is incorrect Suggest checking the quality of the reads prior to performing sequence alignments
54 Read Quality with the FASTX- Toolkit hdp://hannonlab.cshl.edu/fastx_toolkit/
55 Read Quality with the FASTX- Toolkit >fastx_quality_stats i sequence_file.fastq o stats.txt >fastx_quality_boxplot_graph.sh i stats.txt t sample1 o quality.png
56 Read Quality with the FASTX- Toolkit Bad Sequence Good Sequence
57 High Throughput Sequence Alignment TradiAonal sequence alignment algorithms cannot be scaled to align millions of reads
58 High Throughput Sequence Alignment TradiAonal sequence alignment algorithms cannot be scaled to align millions of reads UAlize genome indexing such as Burrows- Wheeler for ultrafast and memory- efficient alignment programs
59 High Throughput Sequence Alignment TradiAonal sequence alignment algorithms cannot be scaled to align millions of reads UAlize genome indexing such as Burrows- Wheeler for ultrafast and memory- efficient alignment programs Next generaaon sequence alignment algorithms are rapidly evolving to accommodate the increasing sequence throughput
60 Tuxedo Suite BowAe fast and quality aware short read aligner for aligning DNA and RNA sequence reads (hdp:// bowae- bio.sourceforge.net/index.shtml) TopHat fast, splice juncaon mapper for RNA- Seq reads built on the BowAe aligner (hdp:// tophat.cbcb.umd.edu/) Cufflinks assembles transcripts, esamates their abundances, and test for differenaal expression and regulaaon using the alignments from BowAe and TopHat
61 BowAe Available for Windows, Mac OS X, Linux, and Solaris
62 BowAe Available for Windows, Mac OS X, Linux, and Solaris Not a general- purpose alignment tool like MUMmer, BLAST, or Vmatch Ideal for aligning short reads to large genomes
63 BowAe Available for Windows, Mac OS X, Linux, and Solaris Not a general- purpose alignment tool like MUMmer, BLAST, or Vmatch Ideal for aligning short reads to large genomes Forms the basis for TopHat, Cufflinks, Crossbow, and Myrna
64 BowAe Available for Windows, Mac OS X, Linux, and Solaris Not a general- purpose alignment tool like MUMmer, BLAST, or Vmatch Ideal for aligning short reads to large genomes Forms the basis for TopHat, Cufflinks, Crossbow, and Myrna Online manual (hdp://bowae- bio.sourceforge.net/manual.shtml) is very helpful to understand the opaons that are available
65 BowAe BowAe Index To print a help screen with the opaonal parameters bowae- build h Command line usage bowae- build [opaons]* <reference_in> <ebwt_base> Example command line bowae- build chr1.fa,chr2.fa,chr3.fa,chr4.fa test_index AddiAonal opaons can be used to improve performance
66 BowAe BowAe Index To print a help screen with the opaonal parameters bowae- build h Command line usage bowae- build [opaons]* <reference_in> <ebwt_base> Example command line bowae- build chr1.fa,chr2.fa,chr3.fa,chr4.fa test_index AddiAonal opaons can be used to improve performance Alignment with BowAe To print a help screen with the opaonal parameters bowae h Command line usage bowae [opaons]* <ebwt> {- 1 <m1> - 2 <m2> <r> <s>} [<hit>] Example command line for single end reads with our test_index BowAe test_index - - solexa1.3- quals test_index sequence_file.fastq > output_file
67 BowAe AddiAonal OpAons: Quality aware MAQ- like mode is the default, BowAe can also be run in a non- quality aware SOAP- like mode BowAe test_index - - solexa1.3- quals v 2 test_index sequence_file.fastq > output_file - v 2 is specifying that there can only be two mismatches in an alignment - a report all valid alignments - k 2 report up to 2 valid alignments - - best report the best alignment - m 2 do not report any alignments for reads with greater than 2 reportable alignments - S print alignments in SAM format which is compaable with SAMtools for subsequent variant calling and manipulaaon of the alignments Many other opaons available
68 TopHat Available for Linux and OS X
69 TopHat Available for Linux and OS X Built on BowAe and uses the same genome index
70 TopHat Available for Linux and OS X Built on BowAe and uses the same genome index Used for alignment of RNA- Seq reads to a genome
71 TopHat Available for Linux and OS X Built on BowAe and uses the same genome index Used for alignment of RNA- Seq reads to a genome OpAmized for paired- end, Illumina sequence reads >70bp
72 TopHat Available for Linux and OS X Built on BowAe and uses the same genome index Used for alignment of RNA- Seq reads to a genome OpAmized for paired- end, Illumina sequence reads >70bp Online manual (hdp://tophat.cbcb.umd.edu/ manual.html) is very helpful to understand the opaons that are available
73 TopHat
74 TopHat Alignment with TopHat To print a help screen with the opaonal parameters tophat h Command line usage tophat [opaons]* <index_base> <reads1_1[,...,readsn_1]> [reads1_2,...readsn_2] Example command line for single end reads with our test_index from before Tophat o output_directory - - solexa1.3- quals test_index sequence_file.fastq AddiAonal opaons - - max- intron size (default is 500kb) - - min- intron size (default is 70bp) - - max- mulahits (default is 40) - - mate- inner- dis (required for paired- end alignment mode) - - num_threads N (runs on N CPUs)
75 TopHat Wiggle tracks can be generated from the TopHat output file accepted_hits.bam Convert to SAM file samtools view h o accepted_hits.sam accepted_hits.bam Generate wiggle file wiggles accepted_hits.sam coverage.wig Wiggle tracks are viewable in programs such as Integrated Genome Browser (hdp://bioviz.org/igb/)
76 AddiAonal Alignment Programs Nature Biotechnology 27, (2009)
77 Valuable Resources
78 QuesAons
Pairwise Sequence Alignment
 Pairwise Sequence Alignment [email protected] SS 2013 Outline Pairwise sequence alignment global - Needleman Wunsch Gotoh algorithm local - Smith Waterman algorithm BLAST - heuristics What
Pairwise Sequence Alignment [email protected] SS 2013 Outline Pairwise sequence alignment global - Needleman Wunsch Gotoh algorithm local - Smith Waterman algorithm BLAST - heuristics What
RETRIEVING SEQUENCE INFORMATION. Nucleotide sequence databases. Database search. Sequence alignment and comparison
 RETRIEVING SEQUENCE INFORMATION Nucleotide sequence databases Database search Sequence alignment and comparison Biological sequence databases Originally just a storage place for sequences. Currently the
RETRIEVING SEQUENCE INFORMATION Nucleotide sequence databases Database search Sequence alignment and comparison Biological sequence databases Originally just a storage place for sequences. Currently the
BLAST. Anders Gorm Pedersen & Rasmus Wernersson
 BLAST Anders Gorm Pedersen & Rasmus Wernersson Database searching Using pairwise alignments to search databases for similar sequences Query sequence Database Database searching Most common use of pairwise
BLAST Anders Gorm Pedersen & Rasmus Wernersson Database searching Using pairwise alignments to search databases for similar sequences Query sequence Database Database searching Most common use of pairwise
Bioinformatics Resources at a Glance
 Bioinformatics Resources at a Glance A Note about FASTA Format There are MANY free bioinformatics tools available online. Bioinformaticists have developed a standard format for nucleotide and protein sequences
Bioinformatics Resources at a Glance A Note about FASTA Format There are MANY free bioinformatics tools available online. Bioinformaticists have developed a standard format for nucleotide and protein sequences
BIOL 3200 Spring 2015 DNA Subway and RNA-Seq Data Analysis
 BIOL 3200 Spring 2015 DNA Subway and RNA-Seq Data Analysis By the end of this lab students should be able to: Describe the uses for each line of the DNA subway program (Red/Yellow/Blue/Green) Describe
BIOL 3200 Spring 2015 DNA Subway and RNA-Seq Data Analysis By the end of this lab students should be able to: Describe the uses for each line of the DNA subway program (Red/Yellow/Blue/Green) Describe
Introduction to NGS data analysis
 Introduction to NGS data analysis Jeroen F. J. Laros Leiden Genome Technology Center Department of Human Genetics Center for Human and Clinical Genetics Sequencing Illumina platforms Characteristics: High
Introduction to NGS data analysis Jeroen F. J. Laros Leiden Genome Technology Center Department of Human Genetics Center for Human and Clinical Genetics Sequencing Illumina platforms Characteristics: High
Databases and mapping BWA. Samtools
 Databases and mapping BWA Samtools FASTQ, SFF, bax.h5 ACE, FASTG FASTA BAM/SAM GFF, BED GenBank/Embl/DDJB many more File formats FASTQ Output format from Illumina and IonTorrent sequencers. Quality scores:
Databases and mapping BWA Samtools FASTQ, SFF, bax.h5 ACE, FASTG FASTA BAM/SAM GFF, BED GenBank/Embl/DDJB many more File formats FASTQ Output format from Illumina and IonTorrent sequencers. Quality scores:
Just the Facts: A Basic Introduction to the Science Underlying NCBI Resources
 1 of 8 11/7/2004 11:00 AM National Center for Biotechnology Information About NCBI NCBI at a Glance A Science Primer Human Genome Resources Model Organisms Guide Outreach and Education Databases and Tools
1 of 8 11/7/2004 11:00 AM National Center for Biotechnology Information About NCBI NCBI at a Glance A Science Primer Human Genome Resources Model Organisms Guide Outreach and Education Databases and Tools
Next Generation Sequencing: Technology, Mapping, and Analysis
 Next Generation Sequencing: Technology, Mapping, and Analysis Gary Benson Computer Science, Biology, Bioinformatics Boston University [email protected] http://tandem.bu.edu/ The Human Genome Project took
Next Generation Sequencing: Technology, Mapping, and Analysis Gary Benson Computer Science, Biology, Bioinformatics Boston University [email protected] http://tandem.bu.edu/ The Human Genome Project took
An example of bioinformatics application on plant breeding projects in Rijk Zwaan
 An example of bioinformatics application on plant breeding projects in Rijk Zwaan Xiangyu Rao 17-08-2012 Introduction of RZ Rijk Zwaan is active worldwide as a vegetable breeding company that focuses on
An example of bioinformatics application on plant breeding projects in Rijk Zwaan Xiangyu Rao 17-08-2012 Introduction of RZ Rijk Zwaan is active worldwide as a vegetable breeding company that focuses on
Version 5.0 Release Notes
 Version 5.0 Release Notes 2011 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Ann Arbor, MI 48108 USA 1.800.497.4939 (USA) +1.734.769.7249 (elsewhere) +1.734.769.7074 (fax) www.genecodes.com
Version 5.0 Release Notes 2011 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Ann Arbor, MI 48108 USA 1.800.497.4939 (USA) +1.734.769.7249 (elsewhere) +1.734.769.7074 (fax) www.genecodes.com
A Tutorial in Genetic Sequence Classification Tools and Techniques
 A Tutorial in Genetic Sequence Classification Tools and Techniques Jake Drew Data Mining CSE 8331 Southern Methodist University [email protected] www.jakemdrew.com Sequence Characters IUPAC nucleotide
A Tutorial in Genetic Sequence Classification Tools and Techniques Jake Drew Data Mining CSE 8331 Southern Methodist University [email protected] www.jakemdrew.com Sequence Characters IUPAC nucleotide
Next generation sequencing (NGS)
") Next generation sequencing (NGS) Vijayachitra Modhukur BIIT [email protected] 1 Bioinformatics course 11/13/12 Sequencing 2 Bioinformatics course 11/13/12 Microarrays vs NGS Sequences do not need to be known
Next generation sequencing (NGS) Vijayachitra Modhukur BIIT [email protected] 1 Bioinformatics course 11/13/12 Sequencing 2 Bioinformatics course 11/13/12 Microarrays vs NGS Sequences do not need to be known
Next Generation Sequencing
 Next Generation Sequencing Technology and applications 10/1/2015 Jeroen Van Houdt - Genomics Core - KU Leuven - UZ Leuven 1 Landmarks in DNA sequencing 1953 Discovery of DNA double helix structure 1977
Next Generation Sequencing Technology and applications 10/1/2015 Jeroen Van Houdt - Genomics Core - KU Leuven - UZ Leuven 1 Landmarks in DNA sequencing 1953 Discovery of DNA double helix structure 1977
Similarity Searches on Sequence Databases: BLAST, FASTA. Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Basel, October 2003
 Similarity Searches on Sequence Databases: BLAST, FASTA Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Basel, October 2003 Outline Importance of Similarity Heuristic Sequence Alignment:
Similarity Searches on Sequence Databases: BLAST, FASTA Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Basel, October 2003 Outline Importance of Similarity Heuristic Sequence Alignment:
Introduction to transcriptome analysis using High Throughput Sequencing technologies (HTS)
") Introduction to transcriptome analysis using High Throughput Sequencing technologies (HTS) A typical RNA Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
Introduction to transcriptome analysis using High Throughput Sequencing technologies (HTS) A typical RNA Seq experiment Library construction Protocol variations Fragmentation methods RNA: nebulization,
Module 1. Sequence Formats and Retrieval. Charles Steward
 The Open Door Workshop Module 1 Sequence Formats and Retrieval Charles Steward 1 Aims Acquaint you with different file formats and associated annotations. Introduce different nucleotide and protein databases.
The Open Door Workshop Module 1 Sequence Formats and Retrieval Charles Steward 1 Aims Acquaint you with different file formats and associated annotations. Introduce different nucleotide and protein databases.
CRAC: An integrated approach to analyse RNA-seq reads Additional File 3 Results on simulated RNA-seq data.
 : An integrated approach to analyse RNA-seq reads Additional File 3 Results on simulated RNA-seq data. Nicolas Philippe and Mikael Salson and Thérèse Commes and Eric Rivals February 13, 2013 1 Results
: An integrated approach to analyse RNA-seq reads Additional File 3 Results on simulated RNA-seq data. Nicolas Philippe and Mikael Salson and Thérèse Commes and Eric Rivals February 13, 2013 1 Results
SMRT Analysis v2.2.0 Overview. 1. SMRT Analysis v2.2.0. 1.1 SMRT Analysis v2.2.0 Overview. Notes:
 SMRT Analysis v2.2.0 Overview 100 338 400 01 1. SMRT Analysis v2.2.0 1.1 SMRT Analysis v2.2.0 Overview Welcome to Pacific Biosciences' SMRT Analysis v2.2.0 Overview 1.2 Contents This module will introduce
SMRT Analysis v2.2.0 Overview 100 338 400 01 1. SMRT Analysis v2.2.0 1.1 SMRT Analysis v2.2.0 Overview Welcome to Pacific Biosciences' SMRT Analysis v2.2.0 Overview 1.2 Contents This module will introduce
Introduction to next-generation sequencing data
 Introduction to next-generation sequencing data David Simpson Centre for Experimental Medicine Queens University Belfast http://www.qub.ac.uk/research-centres/cem/ Outline History of DNA sequencing NGS
Introduction to next-generation sequencing data David Simpson Centre for Experimental Medicine Queens University Belfast http://www.qub.ac.uk/research-centres/cem/ Outline History of DNA sequencing NGS
Gene Models & Bed format: What they represent.
 GeneModels&Bedformat:Whattheyrepresent. Gene models are hypotheses about the structure of transcripts produced by a gene. Like all models, they may be correct, partly correct, or entirely wrong. Typically,
GeneModels&Bedformat:Whattheyrepresent. Gene models are hypotheses about the structure of transcripts produced by a gene. Like all models, they may be correct, partly correct, or entirely wrong. Typically,
Tutorial for Windows and Macintosh. Preparing Your Data for NGS Alignment
 Tutorial for Windows and Macintosh Preparing Your Data for NGS Alignment 2015 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Ann Arbor, MI 48108 USA 1.800.497.4939 (USA) 1.734.769.7249
Tutorial for Windows and Macintosh Preparing Your Data for NGS Alignment 2015 Gene Codes Corporation Gene Codes Corporation 775 Technology Drive, Ann Arbor, MI 48108 USA 1.800.497.4939 (USA) 1.734.769.7249
Next generation DNA sequencing technologies. theory & prac-ce
 Next generation DNA sequencing technologies theory & prac-ce Outline Next- Genera-on sequencing (NGS) technologies overview NGS applica-ons NGS workflow: data collec-on and processing the exome sequencing
Next generation DNA sequencing technologies theory & prac-ce Outline Next- Genera-on sequencing (NGS) technologies overview NGS applica-ons NGS workflow: data collec-on and processing the exome sequencing
Basic processing of next-generation sequencing (NGS) data
 data") Basic processing of next-generation sequencing (NGS) data Getting from raw sequence data to expression analysis! 1 Reminder: we are measuring expression of protein coding genes by transcript abundance
Basic processing of next-generation sequencing (NGS) data Getting from raw sequence data to expression analysis! 1 Reminder: we are measuring expression of protein coding genes by transcript abundance
When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want
 1 When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want to search other databases as well. There are very
1 When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want to search other databases as well. There are very
Using Illumina BaseSpace Apps to Analyze RNA Sequencing Data
 Using Illumina BaseSpace Apps to Analyze RNA Sequencing Data The Illumina TopHat Alignment and Cufflinks Assembly and Differential Expression apps make RNA data analysis accessible to any user, regardless
Using Illumina BaseSpace Apps to Analyze RNA Sequencing Data The Illumina TopHat Alignment and Cufflinks Assembly and Differential Expression apps make RNA data analysis accessible to any user, regardless
CD-HIT User s Guide. Last updated: April 5, 2010. http://cd-hit.org http://bioinformatics.org/cd-hit/
 CD-HIT User s Guide Last updated: April 5, 2010 http://cd-hit.org http://bioinformatics.org/cd-hit/ Program developed by Weizhong Li s lab at UCSD http://weizhong-lab.ucsd.edu [email protected] 1. Introduction
CD-HIT User s Guide Last updated: April 5, 2010 http://cd-hit.org http://bioinformatics.org/cd-hit/ Program developed by Weizhong Li s lab at UCSD http://weizhong-lab.ucsd.edu [email protected] 1. Introduction
Data Processing of Nextera Mate Pair Reads on Illumina Sequencing Platforms
 Data Processing of Nextera Mate Pair Reads on Illumina Sequencing Platforms Introduction Mate pair sequencing enables the generation of libraries with insert sizes in the range of several kilobases (Kb).
Data Processing of Nextera Mate Pair Reads on Illumina Sequencing Platforms Introduction Mate pair sequencing enables the generation of libraries with insert sizes in the range of several kilobases (Kb).
Data Analysis & Management of High-throughput Sequencing Data. Quoclinh Nguyen Research Informatics Genomics Core / Medical Research Institute
 Data Analysis & Management of High-throughput Sequencing Data Quoclinh Nguyen Research Informatics Genomics Core / Medical Research Institute Current Issues Current Issues The QSEQ file Number files per
Data Analysis & Management of High-throughput Sequencing Data Quoclinh Nguyen Research Informatics Genomics Core / Medical Research Institute Current Issues Current Issues The QSEQ file Number files per
Shouguo Gao Ph. D Department of Physics and Comprehensive Diabetes Center
 Computational Challenges in Storage, Analysis and Interpretation of Next-Generation Sequencing Data Shouguo Gao Ph. D Department of Physics and Comprehensive Diabetes Center Next Generation Sequencing
Computational Challenges in Storage, Analysis and Interpretation of Next-Generation Sequencing Data Shouguo Gao Ph. D Department of Physics and Comprehensive Diabetes Center Next Generation Sequencing
Focusing on results not data comprehensive data analysis for targeted next generation sequencing
 Focusing on results not data comprehensive data analysis for targeted next generation sequencing Daniel Swan, Jolyon Holdstock, Angela Matchan, Richard Stark, John Shovelton, Duarte Mohla and Simon Hughes
Focusing on results not data comprehensive data analysis for targeted next generation sequencing Daniel Swan, Jolyon Holdstock, Angela Matchan, Richard Stark, John Shovelton, Duarte Mohla and Simon Hughes
Molecular Databases and Tools
 NWeHealth, The University of Manchester Molecular Databases and Tools Afternoon Session: NCBI/EBI resources, pairwise alignment, BLAST, multiple sequence alignment and primer finding. Dr. Georgina Moulton
NWeHealth, The University of Manchester Molecular Databases and Tools Afternoon Session: NCBI/EBI resources, pairwise alignment, BLAST, multiple sequence alignment and primer finding. Dr. Georgina Moulton
Bioinformatics Grid - Enabled Tools For Biologists.
 Bioinformatics Grid - Enabled Tools For Biologists. What is Grid-Enabled Tools (GET)? As number of data from the genomics and proteomics experiment increases. Problems arise for the current sequence analysis
Bioinformatics Grid - Enabled Tools For Biologists. What is Grid-Enabled Tools (GET)? As number of data from the genomics and proteomics experiment increases. Problems arise for the current sequence analysis
Data formats and file conversions
 Building Excellence in Genomics and Computational Bioscience s Richard Leggett (TGAC) John Walshaw (IFR) Common file formats FASTQ FASTA BAM SAM Raw sequence Alignments MSF EMBL UniProt BED WIG Databases
Building Excellence in Genomics and Computational Bioscience s Richard Leggett (TGAC) John Walshaw (IFR) Common file formats FASTQ FASTA BAM SAM Raw sequence Alignments MSF EMBL UniProt BED WIG Databases
Introduction to Bioinformatics 3. DNA editing and contig assembly
 Introduction to Bioinformatics 3. DNA editing and contig assembly Benjamin F. Matthews United States Department of Agriculture Soybean Genomics and Improvement Laboratory Beltsville, MD 20708 [email protected]
Introduction to Bioinformatics 3. DNA editing and contig assembly Benjamin F. Matthews United States Department of Agriculture Soybean Genomics and Improvement Laboratory Beltsville, MD 20708 [email protected]
Lectures 1 and 8 15. February 7, 2013. Genomics 2012: Repetitorium. Peter N Robinson. VL1: Next- Generation Sequencing. VL8 9: Variant Calling
 Lectures 1 and 8 15 February 7, 2013 This is a review of the material from lectures 1 and 8 14. Note that the material from lecture 15 is not relevant for the final exam. Today we will go over the material
Lectures 1 and 8 15 February 7, 2013 This is a review of the material from lectures 1 and 8 14. Note that the material from lecture 15 is not relevant for the final exam. Today we will go over the material
SGI. High Throughput Computing (HTC) Wrapper Program for Bioinformatics on SGI ICE and SGI UV Systems. January, 2012. Abstract. Haruna Cofer*, PhD
 Wrapper Program for Bioinformatics on SGI ICE and SGI UV Systems. January, 2012. Abstract. Haruna Cofer*, PhD") White Paper SGI High Throughput Computing (HTC) Wrapper Program for Bioinformatics on SGI ICE and SGI UV Systems Haruna Cofer*, PhD January, 2012 Abstract The SGI High Throughput Computing (HTC) Wrapper
White Paper SGI High Throughput Computing (HTC) Wrapper Program for Bioinformatics on SGI ICE and SGI UV Systems Haruna Cofer*, PhD January, 2012 Abstract The SGI High Throughput Computing (HTC) Wrapper
Geospiza s Finch-Server: A Complete Data Management System for DNA Sequencing
 KOO10 5/31/04 12:17 PM Page 131 10 Geospiza s Finch-Server: A Complete Data Management System for DNA Sequencing Sandra Porter, Joe Slagel, and Todd Smith Geospiza, Inc., Seattle, WA Introduction The increased
KOO10 5/31/04 12:17 PM Page 131 10 Geospiza s Finch-Server: A Complete Data Management System for DNA Sequencing Sandra Porter, Joe Slagel, and Todd Smith Geospiza, Inc., Seattle, WA Introduction The increased
Searching Nucleotide Databases
 Searching Nucleotide Databases 1 When we search a nucleic acid databases, Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from the forward strand and 3 reading frames
Searching Nucleotide Databases 1 When we search a nucleic acid databases, Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from the forward strand and 3 reading frames
NGS data analysis. Bernardo J. Clavijo
 NGS data analysis Bernardo J. Clavijo 1 A brief history of DNA sequencing 1953 double helix structure, Watson & Crick! 1977 rapid DNA sequencing, Sanger! 1977 first full (5k) genome bacteriophage Phi X!
NGS data analysis Bernardo J. Clavijo 1 A brief history of DNA sequencing 1953 double helix structure, Watson & Crick! 1977 rapid DNA sequencing, Sanger! 1977 first full (5k) genome bacteriophage Phi X!
A Primer of Genome Science THIRD
 A Primer of Genome Science THIRD EDITION GREG GIBSON-SPENCER V. MUSE North Carolina State University Sinauer Associates, Inc. Publishers Sunderland, Massachusetts USA Contents Preface xi 1 Genome Projects:
A Primer of Genome Science THIRD EDITION GREG GIBSON-SPENCER V. MUSE North Carolina State University Sinauer Associates, Inc. Publishers Sunderland, Massachusetts USA Contents Preface xi 1 Genome Projects:
How Sequencing Experiments Fail
 How Sequencing Experiments Fail v1.0 Simon Andrews [email protected] Classes of Failure Technical Tracking Library Contamination Biological Interpretation Something went wrong with a machine
How Sequencing Experiments Fail v1.0 Simon Andrews [email protected] Classes of Failure Technical Tracking Library Contamination Biological Interpretation Something went wrong with a machine
UGENE Quick Start Guide
 Quick Start Guide This document contains a quick introduction to UGENE. For more detailed information, you can find the UGENE User Manual and other special manuals in project website: http://ugene.unipro.ru.
Quick Start Guide This document contains a quick introduction to UGENE. For more detailed information, you can find the UGENE User Manual and other special manuals in project website: http://ugene.unipro.ru.
Sequence Formats and Sequence Database Searches. Gloria Rendon SC11 Education June, 2011
 Sequence Formats and Sequence Database Searches Gloria Rendon SC11 Education June, 2011 Sequence A is the primary structure of a biological molecule. It is a chain of residues that form a precise linear
Sequence Formats and Sequence Database Searches Gloria Rendon SC11 Education June, 2011 Sequence A is the primary structure of a biological molecule. It is a chain of residues that form a precise linear
3. About R2oDNA Designer
 3. About R2oDNA Designer Please read these publications for more details: Casini A, Christodoulou G, Freemont PS, Baldwin GS, Ellis T, MacDonald JT. R2oDNA Designer: Computational design of biologically-neutral
3. About R2oDNA Designer Please read these publications for more details: Casini A, Christodoulou G, Freemont PS, Baldwin GS, Ellis T, MacDonald JT. R2oDNA Designer: Computational design of biologically-neutral
17 July 2014 WEB-SERVER MANUAL. Contact: Michael Hackenberg ([email protected])
") WEB-SERVER MANUAL Contact: Michael Hackenberg ([email protected]) 1 1 Introduction srnabench is a free web-server tool and standalone application for processing small- RNA data obtained from next generation
WEB-SERVER MANUAL Contact: Michael Hackenberg ([email protected]) 1 1 Introduction srnabench is a free web-server tool and standalone application for processing small- RNA data obtained from next generation
Lecture 4: Exact string searching algorithms. Exact string search algorithms. Definitions. Exact string searching or matching
 COSC 348: Computing for Bioinformatics Definitions A pattern (keyword) is an ordered sequence of symbols. Lecture 4: Exact string searching algorithms Lubica Benuskova http://www.cs.otago.ac.nz/cosc348/
COSC 348: Computing for Bioinformatics Definitions A pattern (keyword) is an ordered sequence of symbols. Lecture 4: Exact string searching algorithms Lubica Benuskova http://www.cs.otago.ac.nz/cosc348/
PreciseTM Whitepaper
 Precise TM Whitepaper Introduction LIMITATIONS OF EXISTING RNA-SEQ METHODS Correctly designed gene expression studies require large numbers of samples, accurate results and low analysis costs. Analysis
Precise TM Whitepaper Introduction LIMITATIONS OF EXISTING RNA-SEQ METHODS Correctly designed gene expression studies require large numbers of samples, accurate results and low analysis costs. Analysis
Biotechnology: DNA Technology & Genomics
 Chapter 20. Biotechnology: DNA Technology & Genomics 2003-2004 The BIG Questions How can we use our knowledge of DNA to: diagnose disease or defect? cure disease or defect? change/improve organisms? What
Chapter 20. Biotechnology: DNA Technology & Genomics 2003-2004 The BIG Questions How can we use our knowledge of DNA to: diagnose disease or defect? cure disease or defect? change/improve organisms? What
BIOINFORMATICS TUTORIAL
 Bio 242 BIOINFORMATICS TUTORIAL Bio 242 α Amylase Lab Sequence Sequence Searches: BLAST Sequence Alignment: Clustal Omega 3d Structure & 3d Alignments DO NOT REMOVE FROM LAB. DO NOT WRITE IN THIS DOCUMENT.
Bio 242 BIOINFORMATICS TUTORIAL Bio 242 α Amylase Lab Sequence Sequence Searches: BLAST Sequence Alignment: Clustal Omega 3d Structure & 3d Alignments DO NOT REMOVE FROM LAB. DO NOT WRITE IN THIS DOCUMENT.
Rapid alignment methods: FASTA and BLAST. p The biological problem p Search strategies p FASTA p BLAST
 Rapid alignment methods: FASTA and BLAST p The biological problem p Search strategies p FASTA p BLAST 257 BLAST: Basic Local Alignment Search Tool p BLAST (Altschul et al., 1990) and its variants are some
Rapid alignment methods: FASTA and BLAST p The biological problem p Search strategies p FASTA p BLAST 257 BLAST: Basic Local Alignment Search Tool p BLAST (Altschul et al., 1990) and its variants are some
Bioinformática BLAST. Blast information guide. Buscas de sequências semelhantes. Search for Homologies BLAST
 BLAST Bioinformática Search for Homologies BLAST BLAST - Basic Local Alignment Search Tool http://blastncbinlmnihgov/blastcgi 1 2 Blast information guide Buscas de sequências semelhantes http://blastncbinlmnihgov/blastcgi?cmd=web&page_type=blastdocs
BLAST Bioinformática Search for Homologies BLAST BLAST - Basic Local Alignment Search Tool http://blastncbinlmnihgov/blastcgi 1 2 Blast information guide Buscas de sequências semelhantes http://blastncbinlmnihgov/blastcgi?cmd=web&page_type=blastdocs
Analysis of ChIP-seq data in Galaxy
 Analysis of ChIP-seq data in Galaxy November, 2012 Local copy: https://galaxy.wi.mit.edu/ Joint project between BaRC and IT Main site: http://main.g2.bx.psu.edu/ 1 Font Conventions Bold and blue refers
Analysis of ChIP-seq data in Galaxy November, 2012 Local copy: https://galaxy.wi.mit.edu/ Joint project between BaRC and IT Main site: http://main.g2.bx.psu.edu/ 1 Font Conventions Bold and blue refers
IOmark Suite. Benchmarking Storage with Applica4on Workloads August, 2013. 2013 Evaluator Group, Inc.
 IOmark Suite Benchmarking Storage with Applica4on Workloads August, 2013 1 What is IOmark Suite?! A storage specific benchmark for applicaaon workloads Tests storage only Supports VDI and Virtual Machine
IOmark Suite Benchmarking Storage with Applica4on Workloads August, 2013 1 What is IOmark Suite?! A storage specific benchmark for applicaaon workloads Tests storage only Supports VDI and Virtual Machine
BioHPC Web Computing Resources at CBSU
 BioHPC Web Computing Resources at CBSU 3CPG workshop Robert Bukowski Computational Biology Service Unit http://cbsu.tc.cornell.edu/lab/doc/biohpc_web_tutorial.pdf BioHPC infrastructure at CBSU BioHPC Web
BioHPC Web Computing Resources at CBSU 3CPG workshop Robert Bukowski Computational Biology Service Unit http://cbsu.tc.cornell.edu/lab/doc/biohpc_web_tutorial.pdf BioHPC infrastructure at CBSU BioHPC Web
Go where the biology takes you. Genome Analyzer IIx Genome Analyzer IIe
 Go where the biology takes you. Genome Analyzer IIx Genome Analyzer IIe Go where the biology takes you. To published results faster With proven scalability To the forefront of discovery To limitless applications
Go where the biology takes you. Genome Analyzer IIx Genome Analyzer IIe Go where the biology takes you. To published results faster With proven scalability To the forefront of discovery To limitless applications
Package hoarder. June 30, 2015
 Type Package Title Information Retrieval for Genetic Datasets Version 0.1 Date 2015-06-29 Author [aut, cre], Anu Sironen [aut] Package hoarder June 30, 2015 Maintainer Depends
Type Package Title Information Retrieval for Genetic Datasets Version 0.1 Date 2015-06-29 Author [aut, cre], Anu Sironen [aut] Package hoarder June 30, 2015 Maintainer Depends
Analysis of NGS Data
 Analysis of NGS Data Introduction and Basics Folie: 1 Overview of Analysis Workflow Images Basecalling Sequences denovo - Sequencing Assembly Annotation Resequencing Alignments Comparison to reference
Analysis of NGS Data Introduction and Basics Folie: 1 Overview of Analysis Workflow Images Basecalling Sequences denovo - Sequencing Assembly Annotation Resequencing Alignments Comparison to reference
SeattleSNPs Interactive Tutorial: Web Tools for Site Selection, Linkage Disequilibrium and Haplotype Analysis
 SeattleSNPs Interactive Tutorial: Web Tools for Site Selection, Linkage Disequilibrium and Haplotype Analysis Goal: This tutorial introduces several websites and tools useful for determining linkage disequilibrium
SeattleSNPs Interactive Tutorial: Web Tools for Site Selection, Linkage Disequilibrium and Haplotype Analysis Goal: This tutorial introduces several websites and tools useful for determining linkage disequilibrium
Note: This document wh_informatics_practical.doc and supporting materials can be downloaded at
 Woods Hole Zebrafish Genetics and Development Bioinformatics/Genomics Lab Ian Woods Note: This document wh_informatics_practical.doc and supporting materials can be downloaded at http://faculty.ithaca.edu/iwoods/docs/wh/
Woods Hole Zebrafish Genetics and Development Bioinformatics/Genomics Lab Ian Woods Note: This document wh_informatics_practical.doc and supporting materials can be downloaded at http://faculty.ithaca.edu/iwoods/docs/wh/
NECC History. Karl V. Steiner 2011 Annual NECC Meeting, Orono, Maine March 15, 2011
 NECC History Karl V. Steiner 2011 Annual NECC Meeting, Orono, Maine March 15, 2011 EPSCoR Cyberinfrastructure Workshop First regional NENI (now NECC) Workshop held in Vermont in August 2007 Workshop heldinkentucky
NECC History Karl V. Steiner 2011 Annual NECC Meeting, Orono, Maine March 15, 2011 EPSCoR Cyberinfrastructure Workshop First regional NENI (now NECC) Workshop held in Vermont in August 2007 Workshop heldinkentucky
Challenges associated with analysis and storage of NGS data
 Challenges associated with analysis and storage of NGS data Gabriella Rustici Research and training coordinator Functional Genomics Group [email protected] Next-generation sequencing Next-generation sequencing
Challenges associated with analysis and storage of NGS data Gabriella Rustici Research and training coordinator Functional Genomics Group [email protected] Next-generation sequencing Next-generation sequencing
The Chinese University of Hong Kong igem 2010 Bacterial based storage and encryp2on device
 The Chinese University of Hong Kong igem 2010 Bacterial based storage and encryp2on device Bacterial based informaaon storage device Bancro4 s group (2001) Mount Sinai School of Meducube Yachie s group
The Chinese University of Hong Kong igem 2010 Bacterial based storage and encryp2on device Bacterial based informaaon storage device Bancro4 s group (2001) Mount Sinai School of Meducube Yachie s group
Genetic Analysis. Phenotype analysis: biological-biochemical analysis. Genotype analysis: molecular and physical analysis
 Genetic Analysis Phenotype analysis: biological-biochemical analysis Behaviour under specific environmental conditions Behaviour of specific genetic configurations Behaviour of progeny in crosses - Genotype
Genetic Analysis Phenotype analysis: biological-biochemical analysis Behaviour under specific environmental conditions Behaviour of specific genetic configurations Behaviour of progeny in crosses - Genotype
Efficient Parallel Execution of Sequence Similarity Analysis Via Dynamic Load Balancing
 Efficient Parallel Execution of Sequence Similarity Analysis Via Dynamic Load Balancing James D. Jackson Philip J. Hatcher Department of Computer Science Kingsbury Hall University of New Hampshire Durham,
Efficient Parallel Execution of Sequence Similarity Analysis Via Dynamic Load Balancing James D. Jackson Philip J. Hatcher Department of Computer Science Kingsbury Hall University of New Hampshire Durham,
8/7/2012. Experimental Design & Intro to NGS Data Analysis. Examples. Agenda. Shoe Example. Breast Cancer Example. Rat Example (Experimental Design)
") Experimental Design & Intro to NGS Data Analysis Ryan Peters Field Application Specialist Partek, Incorporated Agenda Experimental Design Examples ANOVA What assays are possible? NGS Analytical Process
Experimental Design & Intro to NGS Data Analysis Ryan Peters Field Application Specialist Partek, Incorporated Agenda Experimental Design Examples ANOVA What assays are possible? NGS Analytical Process
FlipFlop: Fast Lasso-based Isoform Prediction as a Flow Problem
 FlipFlop: Fast Lasso-based Isoform Prediction as a Flow Problem Elsa Bernard Laurent Jacob Julien Mairal Jean-Philippe Vert September 24, 2013 Abstract FlipFlop implements a fast method for de novo transcript
FlipFlop: Fast Lasso-based Isoform Prediction as a Flow Problem Elsa Bernard Laurent Jacob Julien Mairal Jean-Philippe Vert September 24, 2013 Abstract FlipFlop implements a fast method for de novo transcript
PrimePCR Assay Validation Report
 Gene Information Gene Name Gene Symbol Organism Gene Summary Gene Aliases RefSeq Accession No. UniGene ID Ensembl Gene ID papillary renal cell carcinoma (translocation-associated) PRCC Human This gene
Gene Information Gene Name Gene Symbol Organism Gene Summary Gene Aliases RefSeq Accession No. UniGene ID Ensembl Gene ID papillary renal cell carcinoma (translocation-associated) PRCC Human This gene
Frequently Asked Questions Next Generation Sequencing
 Frequently Asked Questions Next Generation Sequencing Import These Frequently Asked Questions for Next Generation Sequencing are some of the more common questions our customers ask. Questions are divided
Frequently Asked Questions Next Generation Sequencing Import These Frequently Asked Questions for Next Generation Sequencing are some of the more common questions our customers ask. Questions are divided
Design Style of BLAST and FASTA and Their Importance in Human Genome.
 Design Style of BLAST and FASTA and Their Importance in Human Genome. Saba Khalid 1 and Najam-ul-haq 2 SZABIST Karachi, Pakistan Abstract: This subjected study will discuss the concept of BLAST and FASTA.BLAST
Design Style of BLAST and FASTA and Their Importance in Human Genome. Saba Khalid 1 and Najam-ul-haq 2 SZABIST Karachi, Pakistan Abstract: This subjected study will discuss the concept of BLAST and FASTA.BLAST
SeqScape Software Version 2.5 Comprehensive Analysis Solution for Resequencing Applications
 Product Bulletin Sequencing Software SeqScape Software Version 2.5 Comprehensive Analysis Solution for Resequencing Applications Comprehensive reference sequence handling Helps interpret the role of each
Product Bulletin Sequencing Software SeqScape Software Version 2.5 Comprehensive Analysis Solution for Resequencing Applications Comprehensive reference sequence handling Helps interpret the role of each
Biological Databases and Protein Sequence Analysis
 Biological Databases and Protein Sequence Analysis Introduction M. Madan Babu, Center for Biotechnology, Anna University, Chennai 25, India Bioinformatics is the application of Information technology to
Biological Databases and Protein Sequence Analysis Introduction M. Madan Babu, Center for Biotechnology, Anna University, Chennai 25, India Bioinformatics is the application of Information technology to
Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance?
 Optimization 1 Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance? Where to begin? 2 Sequence Databases Swiss-prot MSDB, NCBI nr dbest Species specific ORFS
Optimization 1 Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance? Where to begin? 2 Sequence Databases Swiss-prot MSDB, NCBI nr dbest Species specific ORFS
Exercises for the UCSC Genome Browser Introduction
 Exercises for the UCSC Genome Browser Introduction 1) Find out if the mouse Brca1 gene has non-synonymous SNPs, color them blue, and get external data about a codon-changing SNP. Skills: basic text search;
Exercises for the UCSC Genome Browser Introduction 1) Find out if the mouse Brca1 gene has non-synonymous SNPs, color them blue, and get external data about a codon-changing SNP. Skills: basic text search;
Protein & DNA Sequence Analysis. Bobbie-Jo Webb-Robertson May 3, 2004
 Protein & DNA Sequence Analysis Bobbie-Jo Webb-Robertson May 3, 2004 Sequence Analysis Anything connected to identifying higher biological meaning out of raw sequence data. 2 Genomic & Proteomic Data Sequence
Protein & DNA Sequence Analysis Bobbie-Jo Webb-Robertson May 3, 2004 Sequence Analysis Anything connected to identifying higher biological meaning out of raw sequence data. 2 Genomic & Proteomic Data Sequence
DNA Sequencing Overview
 DNA Sequencing Overview DNA sequencing involves the determination of the sequence of nucleotides in a sample of DNA. It is presently conducted using a modified PCR reaction where both normal and labeled
DNA Sequencing Overview DNA sequencing involves the determination of the sequence of nucleotides in a sample of DNA. It is presently conducted using a modified PCR reaction where both normal and labeled
Hadoopizer : a cloud environment for bioinformatics data analysis
 Hadoopizer : a cloud environment for bioinformatics data analysis Anthony Bretaudeau (1), Olivier Sallou (2), Olivier Collin (3) (1) [email protected], INRIA/Irisa, Campus de Beaulieu, 35042,
Hadoopizer : a cloud environment for bioinformatics data analysis Anthony Bretaudeau (1), Olivier Sallou (2), Olivier Collin (3) (1) [email protected], INRIA/Irisa, Campus de Beaulieu, 35042,
Guide for Bioinformatics Project Module 3
 Structure- Based Evidence and Multiple Sequence Alignment In this module we will revisit some topics we started to look at while performing our BLAST search and looking at the CDD database in the first
Structure- Based Evidence and Multiple Sequence Alignment In this module we will revisit some topics we started to look at while performing our BLAST search and looking at the CDD database in the first
De Novo Assembly Using Illumina Reads
 De Novo Assembly Using Illumina Reads High quality de novo sequence assembly using Illumina Genome Analyzer reads is possible today using publicly available short-read assemblers. Here we summarize the
De Novo Assembly Using Illumina Reads High quality de novo sequence assembly using Illumina Genome Analyzer reads is possible today using publicly available short-read assemblers. Here we summarize the
AS4.1 190509 Replaces 260806 Page 1 of 50 ATF. Software for. DNA Sequencing. Operators Manual. Assign-ATF is intended for Research Use Only (RUO):
:") Replaces 260806 Page 1 of 50 ATF Software for DNA Sequencing Operators Manual Replaces 260806 Page 2 of 50 1 About ATF...5 1.1 Compatibility...5 1.1.1 Computer Operator Systems...5 1.1.2 DNA Sequencing
Replaces 260806 Page 1 of 50 ATF Software for DNA Sequencing Operators Manual Replaces 260806 Page 2 of 50 1 About ATF...5 1.1 Compatibility...5 1.1.1 Computer Operator Systems...5 1.1.2 DNA Sequencing
Nazneen Aziz, PhD. Director, Molecular Medicine Transformation Program Office
 2013 Laboratory Accreditation Program Audioconferences and Webinars Implementing Next Generation Sequencing (NGS) as a Clinical Tool in the Laboratory Nazneen Aziz, PhD Director, Molecular Medicine Transformation
2013 Laboratory Accreditation Program Audioconferences and Webinars Implementing Next Generation Sequencing (NGS) as a Clinical Tool in the Laboratory Nazneen Aziz, PhD Director, Molecular Medicine Transformation
GenBank: A Database of Genetic Sequence Data
 GenBank: A Database of Genetic Sequence Data Computer Science 105 Boston University David G. Sullivan, Ph.D. An Explosion of Scientific Data Scientists are generating ever increasing amounts of data. Relevant
GenBank: A Database of Genetic Sequence Data Computer Science 105 Boston University David G. Sullivan, Ph.D. An Explosion of Scientific Data Scientists are generating ever increasing amounts of data. Relevant
Comparing Methods for Identifying Transcription Factor Target Genes
 Comparing Methods for Identifying Transcription Factor Target Genes Alena van Bömmel (R 3.3.73) Matthew Huska (R 3.3.18) Max Planck Institute for Molecular Genetics Folie 1 Transcriptional Regulation TF
Comparing Methods for Identifying Transcription Factor Target Genes Alena van Bömmel (R 3.3.73) Matthew Huska (R 3.3.18) Max Planck Institute for Molecular Genetics Folie 1 Transcriptional Regulation TF
org.rn.eg.db December 16, 2015 org.rn.egaccnum is an R object that contains mappings between Entrez Gene identifiers and GenBank accession numbers.
 org.rn.eg.db December 16, 2015 org.rn.egaccnum Map Entrez Gene identifiers to GenBank Accession Numbers org.rn.egaccnum is an R object that contains mappings between Entrez Gene identifiers and GenBank
org.rn.eg.db December 16, 2015 org.rn.egaccnum Map Entrez Gene identifiers to GenBank Accession Numbers org.rn.egaccnum is an R object that contains mappings between Entrez Gene identifiers and GenBank
PrimePCR Assay Validation Report
 Gene Information Gene Name sorbin and SH3 domain containing 2 Gene Symbol Organism Gene Summary Gene Aliases RefSeq Accession No. UniGene ID Ensembl Gene ID SORBS2 Human Arg and c-abl represent the mammalian
Gene Information Gene Name sorbin and SH3 domain containing 2 Gene Symbol Organism Gene Summary Gene Aliases RefSeq Accession No. UniGene ID Ensembl Gene ID SORBS2 Human Arg and c-abl represent the mammalian
G E N OM I C S S E RV I C ES
 GENOMICS SERVICES THE NEW YORK GENOME CENTER NYGC is an independent non-profit implementing advanced genomic research to improve diagnosis and treatment of serious diseases. capabilities. N E X T- G E
GENOMICS SERVICES THE NEW YORK GENOME CENTER NYGC is an independent non-profit implementing advanced genomic research to improve diagnosis and treatment of serious diseases. capabilities. N E X T- G E
BIO 3350: ELEMENTS OF BIOINFORMATICS PARTIALLY ONLINE SYLLABUS
 BIO 3350: ELEMENTS OF BIOINFORMATICS PARTIALLY ONLINE SYLLABUS NEW YORK CITY COLLEGE OF TECHNOLOGY The City University Of New York School of Arts and Sciences Biological Sciences Department Course title:
BIO 3350: ELEMENTS OF BIOINFORMATICS PARTIALLY ONLINE SYLLABUS NEW YORK CITY COLLEGE OF TECHNOLOGY The City University Of New York School of Arts and Sciences Biological Sciences Department Course title:
Apply PERL to BioInformatics (II)
") Apply PERL to BioInformatics (II) Lecture Note for Computational Biology 1 (LSM 5191) Jiren Wang http://www.bii.a-star.edu.sg/~jiren BioInformatics Institute Singapore Outline Some examples for manipulating
Apply PERL to BioInformatics (II) Lecture Note for Computational Biology 1 (LSM 5191) Jiren Wang http://www.bii.a-star.edu.sg/~jiren BioInformatics Institute Singapore Outline Some examples for manipulating
RNAseq / ChipSeq / Methylseq and personalized genomics
 RNAseq / ChipSeq / Methylseq and personalized genomics 7711 Lecture Subhajyo) De, PhD Division of Biomedical Informa)cs and Personalized Biomedicine, Department of Medicine University of Colorado School
RNAseq / ChipSeq / Methylseq and personalized genomics 7711 Lecture Subhajyo) De, PhD Division of Biomedical Informa)cs and Personalized Biomedicine, Department of Medicine University of Colorado School
PROC. CAIRO INTERNATIONAL BIOMEDICAL ENGINEERING CONFERENCE 2006 1. E-mail: [email protected]
 BIOINFTool: Bioinformatics and sequence data analysis in molecular biology using Matlab Mai S. Mabrouk 1, Marwa Hamdy 2, Marwa Mamdouh 2, Marwa Aboelfotoh 2,Yasser M. Kadah 2 1 Biomedical Engineering Department,
BIOINFTool: Bioinformatics and sequence data analysis in molecular biology using Matlab Mai S. Mabrouk 1, Marwa Hamdy 2, Marwa Mamdouh 2, Marwa Aboelfotoh 2,Yasser M. Kadah 2 1 Biomedical Engineering Department,
Genomes and SNPs in Malaria and Sickle Cell Anemia
 Genomes and SNPs in Malaria and Sickle Cell Anemia Introduction to Genome Browsing with Ensembl Ensembl The vast amount of information in biological databases today demands a way of organising and accessing
Genomes and SNPs in Malaria and Sickle Cell Anemia Introduction to Genome Browsing with Ensembl Ensembl The vast amount of information in biological databases today demands a way of organising and accessing
Vector NTI Advance 11 Quick Start Guide
 Vector NTI Advance 11 Quick Start Guide Catalog no. 12605050, 12605099, 12605103 Version 11.0 December 15, 2008 12605022 Published by: Invitrogen Corporation 5791 Van Allen Way Carlsbad, CA 92008 U.S.A.
Vector NTI Advance 11 Quick Start Guide Catalog no. 12605050, 12605099, 12605103 Version 11.0 December 15, 2008 12605022 Published by: Invitrogen Corporation 5791 Van Allen Way Carlsbad, CA 92008 U.S.A.
New solutions for Big Data Analysis and Visualization
 New solutions for Big Data Analysis and Visualization From HPC to cloud-based solutions Barcelona, February 2013 Nacho Medina [email protected] http://bioinfo.cipf.es/imedina Head of the Computational Biology
New solutions for Big Data Analysis and Visualization From HPC to cloud-based solutions Barcelona, February 2013 Nacho Medina [email protected] http://bioinfo.cipf.es/imedina Head of the Computational Biology
Module 3. Genome Browsing. Using Web Browsers to View Genome Annota4on. Kers4n Howe Wellcome Trust Sanger Ins4tute zfish- [email protected].
 Module 3 Genome Browsing Using Web Browsers to View Genome Annota4on Kers4n Howe Wellcome Trust Sanger Ins4tute zfish- [email protected] Introduc.on Genome browsing The Ensembl gene set Guided examples
Module 3 Genome Browsing Using Web Browsers to View Genome Annota4on Kers4n Howe Wellcome Trust Sanger Ins4tute zfish- [email protected] Introduc.on Genome browsing The Ensembl gene set Guided examples
A Complete Example of Next- Gen DNA Sequencing Read Alignment. Presentation Title Goes Here
 A Complete Example of Next- Gen DNA Sequencing Read Alignment Presentation Title Goes Here 1 FASTQ Format: The de- facto file format for sharing sequence read data Sequence and a per- base quality score
A Complete Example of Next- Gen DNA Sequencing Read Alignment Presentation Title Goes Here 1 FASTQ Format: The de- facto file format for sharing sequence read data Sequence and a per- base quality score
Discovery and Quantification of RNA with RNASeq Roderic Guigó Serra Centre de Regulació Genòmica (CRG) [email protected]
 roderic.guigo@crg.cat") Bioinformatique et Séquençage Haut Débit, Discovery and Quantification of RNA with RNASeq Roderic Guigó Serra Centre de Regulació Genòmica (CRG) [email protected] 1 RNA Transcription to RNA and subsequent
Bioinformatique et Séquençage Haut Débit, Discovery and Quantification of RNA with RNASeq Roderic Guigó Serra Centre de Regulació Genòmica (CRG) [email protected] 1 RNA Transcription to RNA and subsequent