Bioinformatics Grid - Enabled Tools For Biologists.
|
|
|
- Constance Patrick
- 10 years ago
- Views:
Transcription
1 Bioinformatics Grid - Enabled Tools For Biologists.

2 What is Grid-Enabled Tools (GET)? As number of data from the genomics and proteomics experiment increases. Problems arise for the current sequence analysis technology : mainly SLOWER speed. Using GET the sequence is cut into batches and distributed to different computers in the cluster for processing. After computation, the results are sent back to the head node for recombination and thus is ready for collection by the user. Utilizing this method of sequencing and analyzing data reduces the total amount of time need to be spent in doing so.

3 GET Login Submit sequence in FASTA Format GetANNO GetEMBOSS GetMSA Choose your blast parameter GET Flowchart Choose your parameter Choose to perform either DNA or Protein analysis Blast Emboss Clustalw & Hmmer Results Result in zip is sent via download the zip file

4 GET Click Here to register
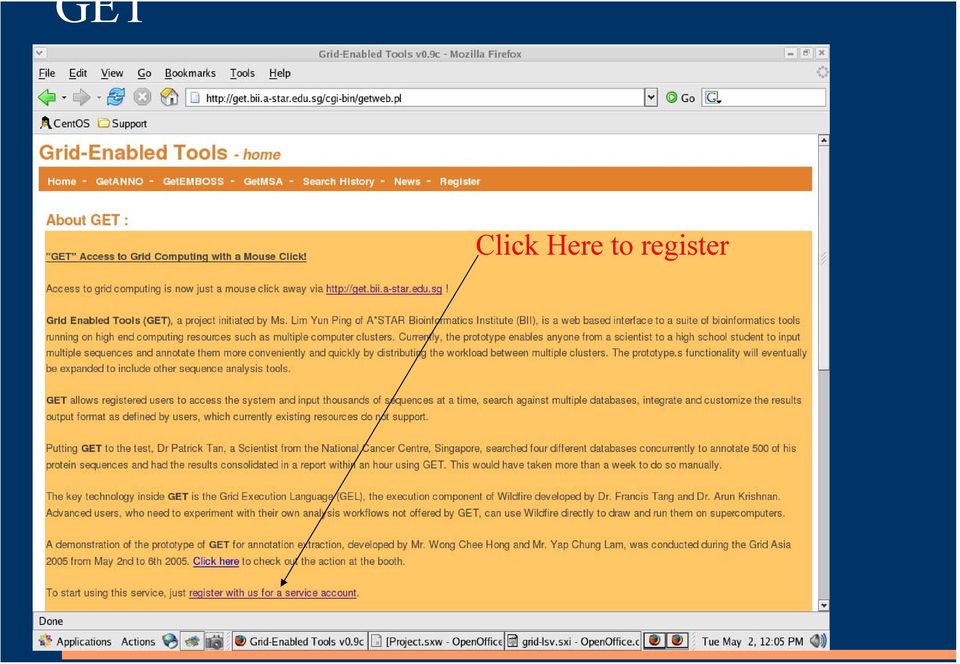
5 Registration Type in your name, and password. Then go to your to activate your account.

6 Login Page Type in your address and password to login

7 GetANNO GetANNO is to add on additional information associated with a particular point in a piece of information. Many proteins are modular in nature, generally many having small conserved regions called motifs. Motifs are surrounded by divergent regions exhibiting a high degree of mutational change among family members of the same protein which tend to correspond to core structural and functional elements of the proteins.

8 GetANNO Protein annotation compares the user input with databases to determine the family of the protein. Computation will take a long time due to large database caused by many classes and long size of proteins. GetANNO splits up the user input into parts and sends it to different computers holding databases to compute, speeding up the time taken to analyze the proteins.

9 GetANNO GetANNO enables users to: - Perform sequence similarity searches against databases such as RefSeq, Swissprot, Pfam and Gene ontology. - Obtain the results description from an excel spreadsheet output.

10 GetANNO Click here to start GetANNO Type in your title Choose which type DNA or Protein Paste in Sequence Choose E-Value Choose type of Matrix Choose the parameter Load Sequence from file Start the Annotation

11 GetANNO Parameter There is 4 types of databases available to BLAST against. There also parameter to choose the E-value and Scoring matrix. In addition a check box is added to only show the top 10 hit in the result

12 Database There is 4 type of database to check against with. RefSeq Gene Ontology Pfam SwissProt All of them are well accurate and reliable since the information is frequently updated.

13 Database RefSeq Provides a comprehensive, integrated & non-redundant set of sequence. Including genomic DNA, transcript (RNA) and protein products. Gene Ontology Provide structured, controlled vocabularies and classification which cover molecular and cellular biology. Often use in annotation of genes, gene products and sequences.

14 Database Pfam A large collection of multiple sequence alignments and hidden Markov model in many common protein domains. SwissProt Provide a high level of annotation, a minimal level of redundancy and high level of integration with other databases.

15 GetEMBOSS EMBOSS collectively contains the processes of: * Sequence alignment * Rapid database searching with sequence patterns * Protein motif identification, including domain analysis * Nucleotide sequence pattern analysis * Codon usage analysis for small genomes * Rapid identification of sequence patterns in large scale sequence sets

16 GetEMBOSS GetEMBOSS helps to save time by splitting up jobs and sent to different computers in the clusters thus the computational power is increased. GetEMBOSS allows users to perform several sequence analysis options on a batch of sequences submitted.

17 GetEMBOSS Click here to start GetEMBOSS Type in your title Paste your FASTA sequence Choose the type of analysis and parameter Load sequence from file Click here to start analysis

18 GetEMBOSS Parameter Find and extract open reading frames. Picks PCR primers and hybridization oligos. Finds restriction enzyme cleavage site. Translates nucleic acid sequence Predicts protein secondary structure Protein statistics Calculates the isoelectric point of a protein Predict transmembrane proteins Predict coiled coil regions

19 GetMSA Multiple Sequence Alignment Compares multiple DNA or amino acid sequences and aligns them to highlight their similarities. GetMSA helps to shorten the computation time needed. Allow users to align multiple sequences for comparison and select further analysis options of predicting secondary structure and finding domains for those regions of interest.

20 GetMSA Click here to start GetMSA Type in your title Choose DNA or Protein sequence Pairwise Alignment options Mutiple Alignment options Type in sequence Load sequence from file Click here to start analysis

21 Search History The Search History is a page where past analysis data done are stored. Results of submitted jobs are found here.
22 Search History Click here to view the result and search history Click here to view the sequence you enter and the result of the analysis
23 Our Project Plans Original Plan NGO BII There is a limited capacity in this system. Often there would be collision between the information travel since it is a single line transmission Users LSF SGE TP Database
24 Linux Virtual Server (LVS) The Linux Virtual Server, or LVS, is a piece of software that is used to balance loads on clusters. The architecture of the whole cluster is transparent to the end user, thus the LVS cluster acts as a single high performance virtual server. LVS is commonly used to build highly scalable services on the internet such as HTTP, FTP, VoIP and so on.
25 Linux Virtual Server (LVS)
26 How LVS Works User Real Server Internet Real Server Load Balancer LAN/WAN Real Server Real Server
27 How LVS Works LVS works by having a load balancer connected to a cluster. The real servers and the load balancer may be interconnected by either high-speed LAN or by geographically dispersed WAN. The load balancer will dispatch requests to the different servers and make parallel services of the cluster to appear as a virtual service on a single IP address, and request dispatching can use IP load balancing technologies or application-level load balancing technologies.
28 How LVS Works Scalability of the system is achieved by transparently adding or removing nodes in the cluster. High availability is provided by detecting node or daemon failures and reconfiguring the system appropriately. Thus, the service will continue to function even if one real server is taken down for maintenance. A backup load balancer can be connected to the network to provide for backup support if the primarily load balancer has gone down due to either maintenance or service failures.
29 How LVS Works
30 How LVS Works can handle >1million concurrent simultaneous connection 128 bytes memory per connection a computer with 1 gigabyte memory can handle more than 8 million simultaneous connections. LVS is also able to produce statistics of each real server, the number of connections, packets, bytes and so on, on which graphs can be created using other software.
31 Our Project Plans Users LVS This is method which make use of a software known as LVS to act as a router to link up all the cluster together. This method is more efficient. NGO BII TP Database synchronized
32 Convention Methods VS GET
33 Start Analysis of 394 Sequences Select Blast parameters Can only submit 1 query sequence at a time. Do not allow upload of file. Repeat the same process for the other 393 sequences. Obtain Results Conventional Blast
34 GetAnno 394 sequence is combined into a single FASTA format text file Start Select Blast parameters Obtain Results Can submit more than 1 query sequence at a time. Allows upload of file.
35 Conventional Blast Time (hr) Vs GetAnno GET Conventional For a 394 sequence, the normal protein blast takes about 18hrs, while GetANNO only takes 2 hours.
36 Conventional Emboss Start Analysis of 10 sequence Can only select 1 Emboss Program Can only submit 1 query sequence at a time. Repeat the same process for the other 9 sequences and also for the other program Obtain Results [Results are not compiled]
37 10 sequence is combined into a Start single FASTA format file Select Emboss Programs [How many depends on user perference] GetEmboss Restrict Running In Parallel Eprimer 3 Can submit more than 1 query sequence at a time. E.g all 10 query seqs Results Results Compile into 1 result text file
38 Conventional Blast Time (mins) Vs GetEmboss GET Conventional For 10 sequence DNA analysis with 2 program, Institute Pasteur Web takes 30mins but Get Emboss takes 2 mins.
39 Conventional MSA Start Upload file that contains more than 1 sequences Choose parameters E.g window size, k-tuple Obtain result [Jalview, alignment, phylogenetic tree] in individual files
40 Start Upload file that contains more than 1 sequence Choose parameters E.g window size, k-tuple GetMSA Allow users the option to build a hmm profile. Obtain result [Jalview, alignment, phylogenetic tree, hmmbuild] in 1 text profile.
41 Conventional MSA Vs GetMSA The GetMSA offers more option of building the hmm profile for their sequence. Thus saving it an extra step
42 Why use our program?? The time taken for GET to complete a process is faster than the conventional method. The GET provide multiple option for analysis. It is more user-friendly than conventional method.
43 Target Audiences Biologists Students Teachers Anyone who need information on DNA or Protein sequencing.
44 Summary Grid Enabled Tools Suite is developed for Biologists to access computing resources via a user friendly web interface for highthroughput bioinformatics analysis. Provide a convenient resource for annotation extraction and sequence analysis Capitalize on the availability of cluster and grid computing to speed up the process.
45 THANK YOU for listening!
SGI. High Throughput Computing (HTC) Wrapper Program for Bioinformatics on SGI ICE and SGI UV Systems. January, 2012. Abstract. Haruna Cofer*, PhD
 Wrapper Program for Bioinformatics on SGI ICE and SGI UV Systems. January, 2012. Abstract. Haruna Cofer*, PhD") White Paper SGI High Throughput Computing (HTC) Wrapper Program for Bioinformatics on SGI ICE and SGI UV Systems Haruna Cofer*, PhD January, 2012 Abstract The SGI High Throughput Computing (HTC) Wrapper
White Paper SGI High Throughput Computing (HTC) Wrapper Program for Bioinformatics on SGI ICE and SGI UV Systems Haruna Cofer*, PhD January, 2012 Abstract The SGI High Throughput Computing (HTC) Wrapper
Guide for Bioinformatics Project Module 3
 Structure- Based Evidence and Multiple Sequence Alignment In this module we will revisit some topics we started to look at while performing our BLAST search and looking at the CDD database in the first
Structure- Based Evidence and Multiple Sequence Alignment In this module we will revisit some topics we started to look at while performing our BLAST search and looking at the CDD database in the first
Linear Sequence Analysis. 3-D Structure Analysis
 Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical properties Molecular weight (MW), isoelectric point (pi), amino acid content, hydropathy (hydrophilic
Linear Sequence Analysis What can you learn from a (single) protein sequence? Calculate it s physical properties Molecular weight (MW), isoelectric point (pi), amino acid content, hydropathy (hydrophilic
Bioinformatics Resources at a Glance
 Bioinformatics Resources at a Glance A Note about FASTA Format There are MANY free bioinformatics tools available online. Bioinformaticists have developed a standard format for nucleotide and protein sequences
Bioinformatics Resources at a Glance A Note about FASTA Format There are MANY free bioinformatics tools available online. Bioinformaticists have developed a standard format for nucleotide and protein sequences
UGENE Quick Start Guide
 Quick Start Guide This document contains a quick introduction to UGENE. For more detailed information, you can find the UGENE User Manual and other special manuals in project website: http://ugene.unipro.ru.
Quick Start Guide This document contains a quick introduction to UGENE. For more detailed information, you can find the UGENE User Manual and other special manuals in project website: http://ugene.unipro.ru.
RETRIEVING SEQUENCE INFORMATION. Nucleotide sequence databases. Database search. Sequence alignment and comparison
 RETRIEVING SEQUENCE INFORMATION Nucleotide sequence databases Database search Sequence alignment and comparison Biological sequence databases Originally just a storage place for sequences. Currently the
RETRIEVING SEQUENCE INFORMATION Nucleotide sequence databases Database search Sequence alignment and comparison Biological sequence databases Originally just a storage place for sequences. Currently the
Biological Databases and Protein Sequence Analysis
 Biological Databases and Protein Sequence Analysis Introduction M. Madan Babu, Center for Biotechnology, Anna University, Chennai 25, India Bioinformatics is the application of Information technology to
Biological Databases and Protein Sequence Analysis Introduction M. Madan Babu, Center for Biotechnology, Anna University, Chennai 25, India Bioinformatics is the application of Information technology to
Bio-Informatics Lectures. A Short Introduction
 Bio-Informatics Lectures A Short Introduction The History of Bioinformatics Sanger Sequencing PCR in presence of fluorescent, chain-terminating dideoxynucleotides Massively Parallel Sequencing Massively
Bio-Informatics Lectures A Short Introduction The History of Bioinformatics Sanger Sequencing PCR in presence of fluorescent, chain-terminating dideoxynucleotides Massively Parallel Sequencing Massively
A Primer of Genome Science THIRD
 A Primer of Genome Science THIRD EDITION GREG GIBSON-SPENCER V. MUSE North Carolina State University Sinauer Associates, Inc. Publishers Sunderland, Massachusetts USA Contents Preface xi 1 Genome Projects:
A Primer of Genome Science THIRD EDITION GREG GIBSON-SPENCER V. MUSE North Carolina State University Sinauer Associates, Inc. Publishers Sunderland, Massachusetts USA Contents Preface xi 1 Genome Projects:
When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want
 1 When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want to search other databases as well. There are very
1 When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want to search other databases as well. There are very
Molecular Databases and Tools
 NWeHealth, The University of Manchester Molecular Databases and Tools Afternoon Session: NCBI/EBI resources, pairwise alignment, BLAST, multiple sequence alignment and primer finding. Dr. Georgina Moulton
NWeHealth, The University of Manchester Molecular Databases and Tools Afternoon Session: NCBI/EBI resources, pairwise alignment, BLAST, multiple sequence alignment and primer finding. Dr. Georgina Moulton
Hidden Markov Models in Bioinformatics. By Máthé Zoltán Kőrösi Zoltán 2006
 Hidden Markov Models in Bioinformatics By Máthé Zoltán Kőrösi Zoltán 2006 Outline Markov Chain HMM (Hidden Markov Model) Hidden Markov Models in Bioinformatics Gene Finding Gene Finding Model Viterbi algorithm
Hidden Markov Models in Bioinformatics By Máthé Zoltán Kőrösi Zoltán 2006 Outline Markov Chain HMM (Hidden Markov Model) Hidden Markov Models in Bioinformatics Gene Finding Gene Finding Model Viterbi algorithm
This document presents the new features available in ngklast release 4.4 and KServer 4.2.
 This document presents the new features available in ngklast release 4.4 and KServer 4.2. 1) KLAST search engine optimization ngklast comes with an updated release of the KLAST sequence comparison tool.
This document presents the new features available in ngklast release 4.4 and KServer 4.2. 1) KLAST search engine optimization ngklast comes with an updated release of the KLAST sequence comparison tool.
BIO 3350: ELEMENTS OF BIOINFORMATICS PARTIALLY ONLINE SYLLABUS
 BIO 3350: ELEMENTS OF BIOINFORMATICS PARTIALLY ONLINE SYLLABUS NEW YORK CITY COLLEGE OF TECHNOLOGY The City University Of New York School of Arts and Sciences Biological Sciences Department Course title:
BIO 3350: ELEMENTS OF BIOINFORMATICS PARTIALLY ONLINE SYLLABUS NEW YORK CITY COLLEGE OF TECHNOLOGY The City University Of New York School of Arts and Sciences Biological Sciences Department Course title:
REGULATIONS FOR THE DEGREE OF BACHELOR OF SCIENCE IN BIOINFORMATICS (BSc[BioInf])
![REGULATIONS FOR THE DEGREE OF BACHELOR OF SCIENCE IN BIOINFORMATICS (BSc[BioInf])](/thumbs/29/13177944.jpg "REGULATIONS FOR THE DEGREE OF BACHELOR OF SCIENCE IN BIOINFORMATICS (BSc[BioInf])") 820 REGULATIONS FOR THE DEGREE OF BACHELOR OF SCIENCE IN BIOINFORMATICS (BSc[BioInf]) (See also General Regulations) BMS1 Admission to the Degree To be eligible for admission to the degree of Bachelor
820 REGULATIONS FOR THE DEGREE OF BACHELOR OF SCIENCE IN BIOINFORMATICS (BSc[BioInf]) (See also General Regulations) BMS1 Admission to the Degree To be eligible for admission to the degree of Bachelor
Just the Facts: A Basic Introduction to the Science Underlying NCBI Resources
 1 of 8 11/7/2004 11:00 AM National Center for Biotechnology Information About NCBI NCBI at a Glance A Science Primer Human Genome Resources Model Organisms Guide Outreach and Education Databases and Tools
1 of 8 11/7/2004 11:00 AM National Center for Biotechnology Information About NCBI NCBI at a Glance A Science Primer Human Genome Resources Model Organisms Guide Outreach and Education Databases and Tools
CD-HIT User s Guide. Last updated: April 5, 2010. http://cd-hit.org http://bioinformatics.org/cd-hit/
 CD-HIT User s Guide Last updated: April 5, 2010 http://cd-hit.org http://bioinformatics.org/cd-hit/ Program developed by Weizhong Li s lab at UCSD http://weizhong-lab.ucsd.edu [email protected] 1. Introduction
CD-HIT User s Guide Last updated: April 5, 2010 http://cd-hit.org http://bioinformatics.org/cd-hit/ Program developed by Weizhong Li s lab at UCSD http://weizhong-lab.ucsd.edu [email protected] 1. Introduction
Biological Sequence Data Formats
 Biological Sequence Data Formats Here we present three standard formats in which biological sequence data (DNA, RNA and protein) can be stored and presented. Raw Sequence: Data without description. FASTA
Biological Sequence Data Formats Here we present three standard formats in which biological sequence data (DNA, RNA and protein) can be stored and presented. Raw Sequence: Data without description. FASTA
Genome Explorer For Comparative Genome Analysis
 Genome Explorer For Comparative Genome Analysis Jenn Conn 1, Jo L. Dicks 1 and Ian N. Roberts 2 Abstract Genome Explorer brings together the tools required to build and compare phylogenies from both sequence
Genome Explorer For Comparative Genome Analysis Jenn Conn 1, Jo L. Dicks 1 and Ian N. Roberts 2 Abstract Genome Explorer brings together the tools required to build and compare phylogenies from both sequence
How To Check If Your Router Is Working Properly On A Nr854T Router (Wnr854) On A Pc Or Mac) On Your Computer Or Ipad (Netbook) On An Ipad Or Ipa (Networking
 On A Pc Or Mac) On Your Computer Or Ipad (Netbook) On An Ipad Or Ipa (Networking") Chapter 7 Using Network Monitoring Tools This chapter describes how to use the maintenance features of your RangeMax NEXT Wireless Router WNR854T. These features can be found by clicking on the Maintenance
Chapter 7 Using Network Monitoring Tools This chapter describes how to use the maintenance features of your RangeMax NEXT Wireless Router WNR854T. These features can be found by clicking on the Maintenance
Lab 2/Phylogenetics/September 16, 2002 1 PHYLOGENETICS
 Lab 2/Phylogenetics/September 16, 2002 1 Read: Tudge Chapter 2 PHYLOGENETICS Objective of the Lab: To understand how DNA and protein sequence information can be used to make comparisons and assess evolutionary
Lab 2/Phylogenetics/September 16, 2002 1 Read: Tudge Chapter 2 PHYLOGENETICS Objective of the Lab: To understand how DNA and protein sequence information can be used to make comparisons and assess evolutionary
UF EDGE brings the classroom to you with online, worldwide course delivery!
 What is the University of Florida EDGE Program? EDGE enables engineering professional, military members, and students worldwide to participate in courses, certificates, and degree programs from the UF
What is the University of Florida EDGE Program? EDGE enables engineering professional, military members, and students worldwide to participate in courses, certificates, and degree programs from the UF
Module 1. Sequence Formats and Retrieval. Charles Steward
 The Open Door Workshop Module 1 Sequence Formats and Retrieval Charles Steward 1 Aims Acquaint you with different file formats and associated annotations. Introduce different nucleotide and protein databases.
The Open Door Workshop Module 1 Sequence Formats and Retrieval Charles Steward 1 Aims Acquaint you with different file formats and associated annotations. Introduce different nucleotide and protein databases.
A Multiple DNA Sequence Translation Tool Incorporating Web Robot and Intelligent Recommendation Techniques
 Proceedings of the 2007 WSEAS International Conference on Computer Engineering and Applications, Gold Coast, Australia, January 17-19, 2007 402 A Multiple DNA Sequence Translation Tool Incorporating Web
Proceedings of the 2007 WSEAS International Conference on Computer Engineering and Applications, Gold Coast, Australia, January 17-19, 2007 402 A Multiple DNA Sequence Translation Tool Incorporating Web
Module 10: Bioinformatics
 Module 10: Bioinformatics 1.) Goal: To understand the general approaches for basic in silico (computer) analysis of DNA- and protein sequences. We are going to discuss sequence formatting required prior
Module 10: Bioinformatics 1.) Goal: To understand the general approaches for basic in silico (computer) analysis of DNA- and protein sequences. We are going to discuss sequence formatting required prior
BUDAPEST: Bioinformatics Utility for Data Analysis of Proteomics using ESTs
 BUDAPEST: Bioinformatics Utility for Data Analysis of Proteomics using ESTs Richard J. Edwards 2008. Contents 1. Introduction... 2 1.1. Version...2 1.2. Using this Manual...2 1.3. Why use BUDAPEST?...2
BUDAPEST: Bioinformatics Utility for Data Analysis of Proteomics using ESTs Richard J. Edwards 2008. Contents 1. Introduction... 2 1.1. Version...2 1.2. Using this Manual...2 1.3. Why use BUDAPEST?...2
Efficient Parallel Execution of Sequence Similarity Analysis Via Dynamic Load Balancing
 Efficient Parallel Execution of Sequence Similarity Analysis Via Dynamic Load Balancing James D. Jackson Philip J. Hatcher Department of Computer Science Kingsbury Hall University of New Hampshire Durham,
Efficient Parallel Execution of Sequence Similarity Analysis Via Dynamic Load Balancing James D. Jackson Philip J. Hatcher Department of Computer Science Kingsbury Hall University of New Hampshire Durham,
Protein & DNA Sequence Analysis. Bobbie-Jo Webb-Robertson May 3, 2004
 Protein & DNA Sequence Analysis Bobbie-Jo Webb-Robertson May 3, 2004 Sequence Analysis Anything connected to identifying higher biological meaning out of raw sequence data. 2 Genomic & Proteomic Data Sequence
Protein & DNA Sequence Analysis Bobbie-Jo Webb-Robertson May 3, 2004 Sequence Analysis Anything connected to identifying higher biological meaning out of raw sequence data. 2 Genomic & Proteomic Data Sequence
Core Bioinformatics. Degree Type Year Semester. 4313473 Bioinformàtica/Bioinformatics OB 0 1
 Core Bioinformatics 2014/2015 Code: 42397 ECTS Credits: 12 Degree Type Year Semester 4313473 Bioinformàtica/Bioinformatics OB 0 1 Contact Name: Sònia Casillas Viladerrams Email: [email protected]
Core Bioinformatics 2014/2015 Code: 42397 ECTS Credits: 12 Degree Type Year Semester 4313473 Bioinformàtica/Bioinformatics OB 0 1 Contact Name: Sònia Casillas Viladerrams Email: [email protected]
How To Build A Clustered Storage Area Network (Csan) From Power All Networks
 From Power All Networks") Power-All Networks Clustered Storage Area Network: A scalable, fault-tolerant, high-performance storage system. Power-All Networks Ltd Abstract: Today's network-oriented computing environments require
Power-All Networks Clustered Storage Area Network: A scalable, fault-tolerant, high-performance storage system. Power-All Networks Ltd Abstract: Today's network-oriented computing environments require
EMBL-EBI Web Services
 EMBL-EBI Web Services Rodrigo Lopez Head of the External Services Team SME Workshop Piemonte 2011 EBI is an Outstation of the European Molecular Biology Laboratory. Summary Introduction The JDispatcher
EMBL-EBI Web Services Rodrigo Lopez Head of the External Services Team SME Workshop Piemonte 2011 EBI is an Outstation of the European Molecular Biology Laboratory. Summary Introduction The JDispatcher
Pairwise Sequence Alignment
 Pairwise Sequence Alignment [email protected] SS 2013 Outline Pairwise sequence alignment global - Needleman Wunsch Gotoh algorithm local - Smith Waterman algorithm BLAST - heuristics What
Pairwise Sequence Alignment [email protected] SS 2013 Outline Pairwise sequence alignment global - Needleman Wunsch Gotoh algorithm local - Smith Waterman algorithm BLAST - heuristics What
Layer 3 Network + Dedicated Internet Connectivity
 Layer 3 Network + Dedicated Internet Connectivity Client: One of the IT Departments in a Northern State Customer's requirement: The customer wanted to establish CAN connectivity (Campus Area Network) for
Layer 3 Network + Dedicated Internet Connectivity Client: One of the IT Departments in a Northern State Customer's requirement: The customer wanted to establish CAN connectivity (Campus Area Network) for
Chapter 6 Using Network Monitoring Tools
 Chapter 6 Using Network Monitoring Tools This chapter describes how to use the maintenance features of your RangeMax Wireless-N Gigabit Router WNR3500. You can access these features by selecting the items
Chapter 6 Using Network Monitoring Tools This chapter describes how to use the maintenance features of your RangeMax Wireless-N Gigabit Router WNR3500. You can access these features by selecting the items
MassMatrix Web Server User Manual
 MassMatrix Web Server User Manual Version 2.2.3 or later Hua Xu, Ph. D. Center for Proteomics & Bioinformatics Case Western Reserve University August 2009 Main Navigation Bar of the Site MassMatrix Web
MassMatrix Web Server User Manual Version 2.2.3 or later Hua Xu, Ph. D. Center for Proteomics & Bioinformatics Case Western Reserve University August 2009 Main Navigation Bar of the Site MassMatrix Web
Activity 7.21 Transcription factors
 Purpose To consolidate understanding of protein synthesis. To explain the role of transcription factors and hormones in switching genes on and off. Play the transcription initiation complex game Regulation
Purpose To consolidate understanding of protein synthesis. To explain the role of transcription factors and hormones in switching genes on and off. Play the transcription initiation complex game Regulation
Searching Nucleotide Databases
 Searching Nucleotide Databases 1 When we search a nucleic acid databases, Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from the forward strand and 3 reading frames
Searching Nucleotide Databases 1 When we search a nucleic acid databases, Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from the forward strand and 3 reading frames
The Lattice Project: A Multi-Model Grid Computing System. Center for Bioinformatics and Computational Biology University of Maryland
 The Lattice Project: A Multi-Model Grid Computing System Center for Bioinformatics and Computational Biology University of Maryland Parallel Computing PARALLEL COMPUTING a form of computation in which
The Lattice Project: A Multi-Model Grid Computing System Center for Bioinformatics and Computational Biology University of Maryland Parallel Computing PARALLEL COMPUTING a form of computation in which
How To Check If Your Router Is Working Properly
 Chapter 6 Using Network Monitoring Tools This chapter describes how to use the maintenance features of your RangeMax Dual Band Wireless-N Router WNDR3300. You can access these features by selecting the
Chapter 6 Using Network Monitoring Tools This chapter describes how to use the maintenance features of your RangeMax Dual Band Wireless-N Router WNDR3300. You can access these features by selecting the
Syllabus of B.Sc. (Bioinformatics) Subject- Bioinformatics (as one subject) B.Sc. I Year Semester I Paper I: Basic of Bioinformatics 85 marks
 Subject- Bioinformatics (as one subject) B.Sc. I Year Semester I Paper I: Basic of Bioinformatics 85 marks") Syllabus of B.Sc. (Bioinformatics) Subject- Bioinformatics (as one subject) B.Sc. I Year Semester I Paper I: Basic of Bioinformatics 85 marks Semester II Paper II: Mathematics I 85 marks B.Sc. II Year
Syllabus of B.Sc. (Bioinformatics) Subject- Bioinformatics (as one subject) B.Sc. I Year Semester I Paper I: Basic of Bioinformatics 85 marks Semester II Paper II: Mathematics I 85 marks B.Sc. II Year
Chapter 6 Using Network Monitoring Tools
 Chapter 6 Using Network Monitoring Tools This chapter describes how to use the maintenance features of your Wireless-G Router Model WGR614v9. You can access these features by selecting the items under
Chapter 6 Using Network Monitoring Tools This chapter describes how to use the maintenance features of your Wireless-G Router Model WGR614v9. You can access these features by selecting the items under
GenBank, Entrez, & FASTA
 GenBank, Entrez, & FASTA Nucleotide Sequence Databases First generation GenBank is a representative example started as sort of a museum to preserve knowledge of a sequence from first discovery great repositories,
GenBank, Entrez, & FASTA Nucleotide Sequence Databases First generation GenBank is a representative example started as sort of a museum to preserve knowledge of a sequence from first discovery great repositories,
Cloud Computing Solutions for Genomics Across Geographic, Institutional and Economic Barriers
 Cloud Computing Solutions for Genomics Across Geographic, Institutional and Economic Barriers Ntinos Krampis Asst. Professor J. Craig Venter Institute [email protected] http://www.jcvi.org/cms/about/bios/kkrampis/
Cloud Computing Solutions for Genomics Across Geographic, Institutional and Economic Barriers Ntinos Krampis Asst. Professor J. Craig Venter Institute [email protected] http://www.jcvi.org/cms/about/bios/kkrampis/
Exercise with Gene Ontology - Cytoscape - BiNGO
 Exercise with Gene Ontology - Cytoscape - BiNGO This practical has material extracted from http://www.cbs.dtu.dk/chipcourse/exercises/ex_go/goexercise11.php In this exercise we will analyze microarray
Exercise with Gene Ontology - Cytoscape - BiNGO This practical has material extracted from http://www.cbs.dtu.dk/chipcourse/exercises/ex_go/goexercise11.php In this exercise we will analyze microarray
Introduction to Bioinformatics AS 250.265 Laboratory Assignment 6
 Introduction to Bioinformatics AS 250.265 Laboratory Assignment 6 In the last lab, you learned how to perform basic multiple sequence alignments. While useful in themselves for determining conserved residues
Introduction to Bioinformatics AS 250.265 Laboratory Assignment 6 In the last lab, you learned how to perform basic multiple sequence alignments. While useful in themselves for determining conserved residues
Sequence Formats and Sequence Database Searches. Gloria Rendon SC11 Education June, 2011
 Sequence Formats and Sequence Database Searches Gloria Rendon SC11 Education June, 2011 Sequence A is the primary structure of a biological molecule. It is a chain of residues that form a precise linear
Sequence Formats and Sequence Database Searches Gloria Rendon SC11 Education June, 2011 Sequence A is the primary structure of a biological molecule. It is a chain of residues that form a precise linear
org.rn.eg.db December 16, 2015 org.rn.egaccnum is an R object that contains mappings between Entrez Gene identifiers and GenBank accession numbers.
 org.rn.eg.db December 16, 2015 org.rn.egaccnum Map Entrez Gene identifiers to GenBank Accession Numbers org.rn.egaccnum is an R object that contains mappings between Entrez Gene identifiers and GenBank
org.rn.eg.db December 16, 2015 org.rn.egaccnum Map Entrez Gene identifiers to GenBank Accession Numbers org.rn.egaccnum is an R object that contains mappings between Entrez Gene identifiers and GenBank
Distributed Data Mining in Discovery Net. Dr. Moustafa Ghanem Department of Computing Imperial College London
 Distributed Data Mining in Discovery Net Dr. Moustafa Ghanem Department of Computing Imperial College London 1. What is Discovery Net 2. Distributed Data Mining for Compute Intensive Tasks 3. Distributed
Distributed Data Mining in Discovery Net Dr. Moustafa Ghanem Department of Computing Imperial College London 1. What is Discovery Net 2. Distributed Data Mining for Compute Intensive Tasks 3. Distributed
Phylogenetic Trees Made Easy
 Phylogenetic Trees Made Easy A How-To Manual Fourth Edition Barry G. Hall University of Rochester, Emeritus and Bellingham Research Institute Sinauer Associates, Inc. Publishers Sunderland, Massachusetts
Phylogenetic Trees Made Easy A How-To Manual Fourth Edition Barry G. Hall University of Rochester, Emeritus and Bellingham Research Institute Sinauer Associates, Inc. Publishers Sunderland, Massachusetts
Introduction to Bioinformatics 2. DNA Sequence Retrieval and comparison
 Introduction to Bioinformatics 2. DNA Sequence Retrieval and comparison Benjamin F. Matthews United States Department of Agriculture Soybean Genomics and Improvement Laboratory Beltsville, MD 20708 [email protected]
Introduction to Bioinformatics 2. DNA Sequence Retrieval and comparison Benjamin F. Matthews United States Department of Agriculture Soybean Genomics and Improvement Laboratory Beltsville, MD 20708 [email protected]
Chapter 2. imapper: A web server for the automated analysis and mapping of insertional mutagenesis sequence data against Ensembl genomes
 Chapter 2. imapper: A web server for the automated analysis and mapping of insertional mutagenesis sequence data against Ensembl genomes 2.1 Introduction Large-scale insertional mutagenesis screening in
Chapter 2. imapper: A web server for the automated analysis and mapping of insertional mutagenesis sequence data against Ensembl genomes 2.1 Introduction Large-scale insertional mutagenesis screening in
Similarity Searches on Sequence Databases: BLAST, FASTA. Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Basel, October 2003
 Similarity Searches on Sequence Databases: BLAST, FASTA Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Basel, October 2003 Outline Importance of Similarity Heuristic Sequence Alignment:
Similarity Searches on Sequence Databases: BLAST, FASTA Lorenza Bordoli Swiss Institute of Bioinformatics EMBnet Course, Basel, October 2003 Outline Importance of Similarity Heuristic Sequence Alignment:
A new type of Hidden Markov Models to predict complex domain architecture in protein sequences
 A new type of Hidden Markov Models to predict complex domain architecture in protein sequences Raluca Uricaru, Laurent Bréhélin and Eric Rivals LIRMM, CNRS Université de Montpellier 2 14 Juin 2007 Raluca
A new type of Hidden Markov Models to predict complex domain architecture in protein sequences Raluca Uricaru, Laurent Bréhélin and Eric Rivals LIRMM, CNRS Université de Montpellier 2 14 Juin 2007 Raluca
Databases and mapping BWA. Samtools
 Databases and mapping BWA Samtools FASTQ, SFF, bax.h5 ACE, FASTG FASTA BAM/SAM GFF, BED GenBank/Embl/DDJB many more File formats FASTQ Output format from Illumina and IonTorrent sequencers. Quality scores:
Databases and mapping BWA Samtools FASTQ, SFF, bax.h5 ACE, FASTG FASTA BAM/SAM GFF, BED GenBank/Embl/DDJB many more File formats FASTQ Output format from Illumina and IonTorrent sequencers. Quality scores:
When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want
 1 When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want to search other databases as well. There are very
1 When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want to search other databases as well. There are very
SNMP and Web-based Load Cluster Management System
 and Web-based Load Cluster Management System Myungsup Kim and J. Won-Ki Hong Distributed Processing & Network Management Lab. Dept. of Computer Science and Engineering, Pohang Korea Tel: +82-54-279-5654
and Web-based Load Cluster Management System Myungsup Kim and J. Won-Ki Hong Distributed Processing & Network Management Lab. Dept. of Computer Science and Engineering, Pohang Korea Tel: +82-54-279-5654
SICKLE CELL ANEMIA & THE HEMOGLOBIN GENE TEACHER S GUIDE
 AP Biology Date SICKLE CELL ANEMIA & THE HEMOGLOBIN GENE TEACHER S GUIDE LEARNING OBJECTIVES Students will gain an appreciation of the physical effects of sickle cell anemia, its prevalence in the population,
AP Biology Date SICKLE CELL ANEMIA & THE HEMOGLOBIN GENE TEACHER S GUIDE LEARNING OBJECTIVES Students will gain an appreciation of the physical effects of sickle cell anemia, its prevalence in the population,
2.3 Identify rrna sequences in DNA
 2.3 Identify rrna sequences in DNA For identifying rrna sequences in DNA we will use rnammer, a program that implements an algorithm designed to find rrna sequences in DNA [5]. The program was made by
2.3 Identify rrna sequences in DNA For identifying rrna sequences in DNA we will use rnammer, a program that implements an algorithm designed to find rrna sequences in DNA [5]. The program was made by
In Memory Accelerator for MongoDB
 In Memory Accelerator for MongoDB Yakov Zhdanov, Director R&D GridGain Systems GridGain: In Memory Computing Leader 5 years in production 100s of customers & users Starts every 10 secs worldwide Over 15,000,000
In Memory Accelerator for MongoDB Yakov Zhdanov, Director R&D GridGain Systems GridGain: In Memory Computing Leader 5 years in production 100s of customers & users Starts every 10 secs worldwide Over 15,000,000
PROC. CAIRO INTERNATIONAL BIOMEDICAL ENGINEERING CONFERENCE 2006 1. E-mail: [email protected]
 BIOINFTool: Bioinformatics and sequence data analysis in molecular biology using Matlab Mai S. Mabrouk 1, Marwa Hamdy 2, Marwa Mamdouh 2, Marwa Aboelfotoh 2,Yasser M. Kadah 2 1 Biomedical Engineering Department,
BIOINFTool: Bioinformatics and sequence data analysis in molecular biology using Matlab Mai S. Mabrouk 1, Marwa Hamdy 2, Marwa Mamdouh 2, Marwa Aboelfotoh 2,Yasser M. Kadah 2 1 Biomedical Engineering Department,
Chapter 5. Data Communication And Internet Technology
 Chapter 5 Data Communication And Internet Technology Purpose Understand the fundamental networking concepts Agenda Network Concepts Communication Protocol TCP/IP-OSI Architecture Network Types LAN WAN
Chapter 5 Data Communication And Internet Technology Purpose Understand the fundamental networking concepts Agenda Network Concepts Communication Protocol TCP/IP-OSI Architecture Network Types LAN WAN
BLAST. Anders Gorm Pedersen & Rasmus Wernersson
 BLAST Anders Gorm Pedersen & Rasmus Wernersson Database searching Using pairwise alignments to search databases for similar sequences Query sequence Database Database searching Most common use of pairwise
BLAST Anders Gorm Pedersen & Rasmus Wernersson Database searching Using pairwise alignments to search databases for similar sequences Query sequence Database Database searching Most common use of pairwise
3. About R2oDNA Designer
 3. About R2oDNA Designer Please read these publications for more details: Casini A, Christodoulou G, Freemont PS, Baldwin GS, Ellis T, MacDonald JT. R2oDNA Designer: Computational design of biologically-neutral
3. About R2oDNA Designer Please read these publications for more details: Casini A, Christodoulou G, Freemont PS, Baldwin GS, Ellis T, MacDonald JT. R2oDNA Designer: Computational design of biologically-neutral
BioHPC Web Computing Resources at CBSU
 BioHPC Web Computing Resources at CBSU 3CPG workshop Robert Bukowski Computational Biology Service Unit http://cbsu.tc.cornell.edu/lab/doc/biohpc_web_tutorial.pdf BioHPC infrastructure at CBSU BioHPC Web
BioHPC Web Computing Resources at CBSU 3CPG workshop Robert Bukowski Computational Biology Service Unit http://cbsu.tc.cornell.edu/lab/doc/biohpc_web_tutorial.pdf BioHPC infrastructure at CBSU BioHPC Web
The sequence of bases on the mrna is a code that determines the sequence of amino acids in the polypeptide being synthesized:
 Module 3F Protein Synthesis So far in this unit, we have examined: How genes are transmitted from one generation to the next Where genes are located What genes are made of How genes are replicated How
Module 3F Protein Synthesis So far in this unit, we have examined: How genes are transmitted from one generation to the next Where genes are located What genes are made of How genes are replicated How
REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])
![REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])](/thumbs/33/16674036.jpg "REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])") 305 REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc]) (See also General Regulations) Any publication based on work approved for a higher degree should contain a reference
305 REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc]) (See also General Regulations) Any publication based on work approved for a higher degree should contain a reference
A Web Based Software for Synonymous Codon Usage Indices
 International Journal of Information and Computation Technology. ISSN 0974-2239 Volume 3, Number 3 (2013), pp. 147-152 International Research Publications House http://www. irphouse.com /ijict.htm A Web
International Journal of Information and Computation Technology. ISSN 0974-2239 Volume 3, Number 3 (2013), pp. 147-152 International Research Publications House http://www. irphouse.com /ijict.htm A Web
Focusing on results not data comprehensive data analysis for targeted next generation sequencing
 Focusing on results not data comprehensive data analysis for targeted next generation sequencing Daniel Swan, Jolyon Holdstock, Angela Matchan, Richard Stark, John Shovelton, Duarte Mohla and Simon Hughes
Focusing on results not data comprehensive data analysis for targeted next generation sequencing Daniel Swan, Jolyon Holdstock, Angela Matchan, Richard Stark, John Shovelton, Duarte Mohla and Simon Hughes
Apply PERL to BioInformatics (II)
") Apply PERL to BioInformatics (II) Lecture Note for Computational Biology 1 (LSM 5191) Jiren Wang http://www.bii.a-star.edu.sg/~jiren BioInformatics Institute Singapore Outline Some examples for manipulating
Apply PERL to BioInformatics (II) Lecture Note for Computational Biology 1 (LSM 5191) Jiren Wang http://www.bii.a-star.edu.sg/~jiren BioInformatics Institute Singapore Outline Some examples for manipulating
IEEE International Conference on Computing, Analytics and Security Trends CAST-2016 (19 21 December, 2016) Call for Paper
 Call for Paper") IEEE International Conference on Computing, Analytics and Security Trends CAST-2016 (19 21 December, 2016) Call for Paper CAST-2015 provides an opportunity for researchers, academicians, scientists and
IEEE International Conference on Computing, Analytics and Security Trends CAST-2016 (19 21 December, 2016) Call for Paper CAST-2015 provides an opportunity for researchers, academicians, scientists and
A Tutorial in Genetic Sequence Classification Tools and Techniques
 A Tutorial in Genetic Sequence Classification Tools and Techniques Jake Drew Data Mining CSE 8331 Southern Methodist University [email protected] www.jakemdrew.com Sequence Characters IUPAC nucleotide
A Tutorial in Genetic Sequence Classification Tools and Techniques Jake Drew Data Mining CSE 8331 Southern Methodist University [email protected] www.jakemdrew.com Sequence Characters IUPAC nucleotide
BIOINFORMATICS TUTORIAL
 Bio 242 BIOINFORMATICS TUTORIAL Bio 242 α Amylase Lab Sequence Sequence Searches: BLAST Sequence Alignment: Clustal Omega 3d Structure & 3d Alignments DO NOT REMOVE FROM LAB. DO NOT WRITE IN THIS DOCUMENT.
Bio 242 BIOINFORMATICS TUTORIAL Bio 242 α Amylase Lab Sequence Sequence Searches: BLAST Sequence Alignment: Clustal Omega 3d Structure & 3d Alignments DO NOT REMOVE FROM LAB. DO NOT WRITE IN THIS DOCUMENT.
REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])
![REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])](/thumbs/25/6066281.jpg "REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])") 299 REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc]) (See also General Regulations) Any publication based on work approved for a higher degree should contain a reference
299 REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc]) (See also General Regulations) Any publication based on work approved for a higher degree should contain a reference
Agenda. Distributed System Structures. Why Distributed Systems? Motivation
 Agenda Distributed System Structures CSCI 444/544 Operating Systems Fall 2008 Motivation Network structure Fundamental network services Sockets and ports Client/server model Remote Procedure Call (RPC)
Agenda Distributed System Structures CSCI 444/544 Operating Systems Fall 2008 Motivation Network structure Fundamental network services Sockets and ports Client/server model Remote Procedure Call (RPC)
Quick Start Guide. Cerberus FTP is distributed in Canada through C&C Software. Visit us today at www.ccsoftware.ca!
 Quick Start Guide Cerberus FTP is distributed in Canada through C&C Software. Visit us today at www.ccsoftware.ca! How to Setup a File Server with Cerberus FTP Server FTP and SSH SFTP are application protocols
Quick Start Guide Cerberus FTP is distributed in Canada through C&C Software. Visit us today at www.ccsoftware.ca! How to Setup a File Server with Cerberus FTP Server FTP and SSH SFTP are application protocols
EMBOSS A data analysis package
 EMBOSS A data analysis package Adapted from course developed by Lisa Mullin (EMBL-EBI) and David Judge Cambridge University EMBOSS is a free Open Source software analysis package specially developed for
EMBOSS A data analysis package Adapted from course developed by Lisa Mullin (EMBL-EBI) and David Judge Cambridge University EMBOSS is a free Open Source software analysis package specially developed for
PARALLEL & CLUSTER COMPUTING CS 6260 PROFESSOR: ELISE DE DONCKER BY: LINA HUSSEIN
 1 PARALLEL & CLUSTER COMPUTING CS 6260 PROFESSOR: ELISE DE DONCKER BY: LINA HUSSEIN Introduction What is cluster computing? Classification of Cluster Computing Technologies: Beowulf cluster Construction
1 PARALLEL & CLUSTER COMPUTING CS 6260 PROFESSOR: ELISE DE DONCKER BY: LINA HUSSEIN Introduction What is cluster computing? Classification of Cluster Computing Technologies: Beowulf cluster Construction
Translation Study Guide
 Translation Study Guide This study guide is a written version of the material you have seen presented in the replication unit. In translation, the cell uses the genetic information contained in mrna to
Translation Study Guide This study guide is a written version of the material you have seen presented in the replication unit. In translation, the cell uses the genetic information contained in mrna to
Unipro UGENE User Manual Version 1.12.3
 Unipro UGENE User Manual Version 1.12.3 April 01, 2014 Contents 1 About Unipro................................... 10 1.1 Contacts.......................................... 10 2 About UGENE..................................
Unipro UGENE User Manual Version 1.12.3 April 01, 2014 Contents 1 About Unipro................................... 10 1.1 Contacts.......................................... 10 2 About UGENE..................................
Current Motif Discovery Tools and their Limitations
 Current Motif Discovery Tools and their Limitations Philipp Bucher SIB / CIG Workshop 3 October 2006 Trendy Concepts and Hypotheses Transcription regulatory elements act in a context-dependent manner.
Current Motif Discovery Tools and their Limitations Philipp Bucher SIB / CIG Workshop 3 October 2006 Trendy Concepts and Hypotheses Transcription regulatory elements act in a context-dependent manner.
Leased Line + Remote Dial-in connectivity
 Leased Line + Remote Dial-in connectivity Client: One of the TELCO offices in a Southern state. The customer wanted to establish WAN Connectivity between central location and 10 remote locations. The customer
Leased Line + Remote Dial-in connectivity Client: One of the TELCO offices in a Southern state. The customer wanted to establish WAN Connectivity between central location and 10 remote locations. The customer
REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])
![REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])](/thumbs/24/2595933.jpg "REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc])") 244 REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc]) (See also General Regulations) Any publication based on work approved for a higher degree should contain a reference
244 REGULATIONS FOR THE DEGREE OF MASTER OF SCIENCE IN COMPUTER SCIENCE (MSc[CompSc]) (See also General Regulations) Any publication based on work approved for a higher degree should contain a reference
Cisco WAAS for Isilon IQ
 Cisco WAAS for Isilon IQ Integrating Cisco WAAS with Isilon IQ Clustered Storage to Enable the Next-Generation Data Center An Isilon Systems/Cisco Systems Whitepaper January 2008 1 Table of Contents 1.
Cisco WAAS for Isilon IQ Integrating Cisco WAAS with Isilon IQ Clustered Storage to Enable the Next-Generation Data Center An Isilon Systems/Cisco Systems Whitepaper January 2008 1 Table of Contents 1.
LabGenius. Technical design notes. The world s most advanced synthetic DNA libraries. [email protected] V1.5 NOV 15
 LabGenius The world s most advanced synthetic DNA libraries Technical design notes [email protected] V1.5 NOV 15 Introduction OUR APPROACH LabGenius is a gene synthesis company focussed on the design and manufacture
LabGenius The world s most advanced synthetic DNA libraries Technical design notes [email protected] V1.5 NOV 15 Introduction OUR APPROACH LabGenius is a gene synthesis company focussed on the design and manufacture
The Steps. 1. Transcription. 2. Transferal. 3. Translation
 Protein Synthesis Protein synthesis is simply the "making of proteins." Although the term itself is easy to understand, the multiple steps that a cell in a plant or animal must go through are not. In order
Protein Synthesis Protein synthesis is simply the "making of proteins." Although the term itself is easy to understand, the multiple steps that a cell in a plant or animal must go through are not. In order
EMBL Identity & Access Management
 EMBL Identity & Access Management Rupert Lück EMBL Heidelberg e IRG Workshop Zürich Apr 24th 2008 Outline EMBL Overview Identity & Access Management for EMBL IT Requirements & Strategy Project Goal and
EMBL Identity & Access Management Rupert Lück EMBL Heidelberg e IRG Workshop Zürich Apr 24th 2008 Outline EMBL Overview Identity & Access Management for EMBL IT Requirements & Strategy Project Goal and
Library page. SRS first view. Different types of database in SRS. Standard query form
 SRS & Entrez SRS Sequence Retrieval System Bengt Persson Whatis SRS? Sequence Retrieval System User-friendly interface to databases http://srs.ebi.ac.uk Developed by Thure Etzold and co-workers EMBL/EBI
SRS & Entrez SRS Sequence Retrieval System Bengt Persson Whatis SRS? Sequence Retrieval System User-friendly interface to databases http://srs.ebi.ac.uk Developed by Thure Etzold and co-workers EMBL/EBI
CPAS Overview. Josh Eckels LabKey Software [email protected]
 CPAS Overview Josh Eckels LabKey Software [email protected] CPAS Web-based system for processing, storing, and analyzing results of MS/MS experiments Key goals: Provide a great analysis front-end for
CPAS Overview Josh Eckels LabKey Software [email protected] CPAS Web-based system for processing, storing, and analyzing results of MS/MS experiments Key goals: Provide a great analysis front-end for