The information contained in these slides is for general purposes only and presents the state of knowledge at November 30, 2011
|
|
|
- Ilene Jefferson
- 8 years ago
- Views:
Transcription
1 1
2 The information contained in these slides is for general purposes only and presents the state of knowledge at November 30, 2011 No rights can be derived from this information The Medicines Evaluation Board accepts no liability for direct or consequential damage resulting from teh use of, reliance on or action taken on the basis of this information provided during this session 2

3 Better protecting the patients Be aware of the system and your role

4 Overzicht presentatie Introductie,doel van de veranderingen Wetgeving Verordening Richtlijn Implementatie maatregelen Belangrijke veranderingen in vogelvlucht Inleiding op de nieuwe wetgeving 4

5 Pharmacovigilantie Spontaneous reports 2001 cerivastatine 2004 Vioxx 1995 PSURs RMPs 2006 EU consultation

6

7

8 Pharmacovigilantie Spontaneous reports 2001 cerivastatine 2004 Vioxx 1995 PSURs RMPs 2010 Regulation Directive 2006 EU consultation

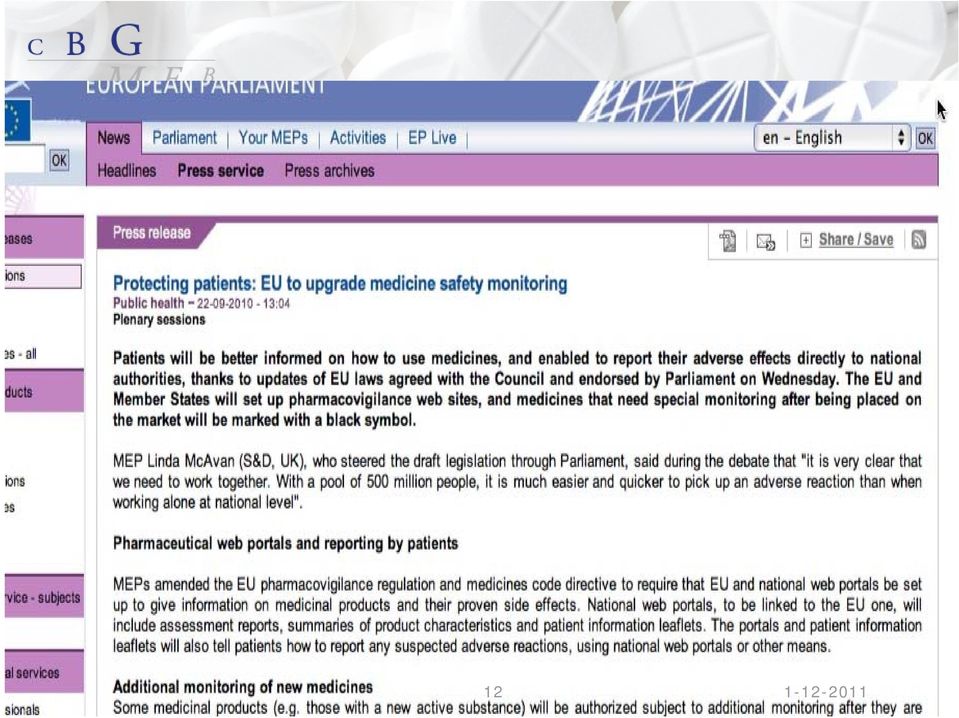
9 European Commission schatting 5% of all hospital admissions due to ADRs 5% of all hospital patients experience an ADR ADRs5 th most common cause of hospital death 197,000 deaths per year in EU caused by ADRs Total societal cost 79 billion

10 10

11 Aims of the new legislation Clear roles and responsibilities Integrate benefit and risks Science based ( embrace the hierarchy of evidence) Risk based /proportionate Increased planning/proactivity Ensure robust and rapid decision making Strengthening the EU network Engage patients and HCPs Increase transparancy and Reduce accountability duplication/redundancy Provide better information on medicines

12 12
13 Overzicht presentatie Introductie,doel van de veranderingen Wetgeving Verordening Richtlijn Implementatie maatregelen Belangrijke veranderingen in vogelvlucht 13

14 Regulation/verordening 1235/2010 Focus op EU procedures en de taken van EMA Directive/richtlijn 2010/84/EC Focus op farmacovigilantie activiteiten in de lidstaten met het oog op nationale registraties Maatregelen gelden voor de centrale (CAPs) en niet centrale producten (non CAPS) Moeten samen gelezen worden, beiden treden in werking juli
 Moeten samen gelezen worden, beiden treden in")
15 15

16 Implementing measures EC EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Health systems and products Pharmaceuticals Brussels, SANCO/D3/FS/cg/ddg1.d.3(2011) IMPLEMENTING MEASURES IN ORDER TO HARMONISE THE PERFORMANCE OF THE PHARMACOVIGILANCE ACTIVITIES PROVIDED FOR IN DIRECTIVE 2001/83/EC AND REGULATION (EC) NO 726/2004 THE CONCEPT PAPER SUBMITTED FOR PUBLIC CONSULTATION Deadline for Public Consultation: 7 November 2011 This document does not represent an official position of the European Commission. It is a tool to explore the views of interested parties on a preliminary draft. The suggestions contained in this document do not prejudge the form and content of any future proposal by the European Commission. 16

17 Implementing measures EC Pharmacovigilance system master file Quality systems for the performance of pharmacovigilance activities Common obligations Quality systems for the performance of pharmacovigilance activities by marketing authorisation holders Quality systems for the performance of pharmacovigilance activities by national competent authorities and EMA Signal detection and risk identification Use of terminology Transmission and Submission requirements 17

18 Implementing measures EC G. Transmission and Submission requirements Annex I Electronic submissions of suspected adverse reactions Annex II Risk management plans Annex III Electronic periodic safety update reports Annex IV Protocols, abstracts and final study reports for the post-authorisation safety studies 18

19 Overzicht presentatie Introductie,doel van de veranderingen Wetgeving Verordening Richtlijn Implementatie maatregelen Belangrijke veranderingen in vogelvlucht 19

20 Wat gaat de wet veranderen? Risk management plan Literatuur monitoring Additional monitoring Referrals union procedures PSURs Post authorisation safety studies renewal Bijwerkingen Meten effecten risk minimisation Wetenschappelijke committees /PRAC Webportals Farmacovigilantie master file Farmacovigilantie inspecties Transparantie 20 Signaal detectie Worksharing

21 Overzicht presentatie Introductie,doel van de veranderingen Wetgeving Verordening Richtlijn Implementatie maatregelen Belangrijke veranderingen in vogelvlucht PRAC PASS PhV MF Additional monitoring 21
22 Pharmacovigilance Risk Assessment Committee (PRAC) Mandate (R61a) All aspects of the risk management of the use of medicinal products including the detection, assessment, minimisation and communication relating to the risk of adverse reactions, having due regard to the therapeutic effect of the medicinal product, the design and evaluation of post-authorisation safety studies and pharmacovigilance audit 222 2
23 23
24 24
25 25
26 Besluitvormings model PSUR PASS Union procedures (art. 31, 107i) RMP CMD(h) (no CAP) PRAC recommendation: regulatory action CHMP (at least 1 CAP) Position = maint., var., susp., revoc. Opinion = maint., var., susp., revoc. If consensus: agreement NO consensus: position majority COMMISSION MS: adopt measures MAH: submit variation Decision addressed to MS Decision modifying MA (CAP)
27 Overzicht presentatie Introductie,doel van de veranderingen Wetgeving Verordening Richtlijn Implementatie maatregelen Belangrijke veranderingen in vogelvlucht PRAC PASS PhVS-MF Additional monitoring 27
28 PASS Post-authorisation Safety Studies (PASS) (1/4) Strengthened legal basis Regulators can require PASS at first authorisation Regulators can require PASS post-authorisation PASS is a condition of the authorisation and is legally binding In the event that the same safety concern applies to more than one medicinal product, the EMA/national competent authority shall encourage the marketing authorisation holders concerned to conduct a joint post-authorisation safety study 28
29 PASS Post-authorisation Safety Studies (PASS) (2/4) Principles for and oversight of PASS Definition: Principles and oversight for non-interventional PASS which are: Any study relating to an authorised medicinal product conducted with the aim of identifying, characterising or quantifying a safety hazard, confirming the safety profile of the medicinal product, or of measuring the effectiveness of risk management measures initiated, managed or financed by the MAH voluntarily or pursuant to obligations imposed [by regulators] and which involve the collection of data on suspected adverse reactions from patients or healthcare professionals. Still may have additional MS requirements on well being and rights of participants in non-interventional PASS 29
30 PASS ALL PASS ALL PASS Initiated, managed or financed by MAH, involving data collection from patients or HCPs PASS Required by regulators PASS In more than one MS 30
31 PASS Post-authorisation Safety Studies (PASS) (3/4) Principles for and oversight of all PASS (initiated, managed or financed by MAH) The studies shall not be performed where the act of conducting the study promotes the use of a medicinal product. Payments to healthcare professionals for participating in noninterventional PASS shall be restricted to the compensation for time and expenses incurred. National competent authority may require the MAH to submit the protocol and the progress reports to the competent authorities of the MSs in which the study is conducted. MAH shall send the final report to the competent authorities of the MSs in which the study was conducted within 12 months of the end of data collection. Any new information which might influence the evaluation of the riskbenefit balance of the medicinal product shall be communicated [by company] to the competent authorities of the MS in which the product has been authorised. 31
32 PASS Post-authorisation Safety Studies (PASS) (4/4) Principles for and oversight of PASS for studies required by regulators Protocols actively approved prior to study (single country = MS, multiple country = PRAC) Protocol amendments actively approved Final report and abstract submitted Automatic, formal assessment and decision-making based on results Company updates product information 32
33 PASS ALL PASS ALL PASS Initiated, managed or financed by MAH, involving data collection from patients or HCPs PASS Required by regulators PASS In more than one MS 33
34 Overzicht presentatie Introductie,doel van de veranderingen Wetgeving Verordening Richtlijn Implementatie maatregelen Belangrijke veranderingen in vogelvlucht PRAC PASS PhVS MF Additional monitoring 34
35 DDPS- PhVS master file 35
36 DDPS- PhVS master file The marketing authorisation holder should have: a pharmacovigilance system ensure the monitoring and supervision of authorised medicinal products recorded in a pharmacovigilance system master file permanently available for inspection (request CA). Applications for marketing authorisations should therefore be accompanied by a brief description of the corresponding pharmacovigilance system, location where the pharmacovigilance system master file is kept QPPV 36
37 DDPS- PhVS master file Pharmacovigilance system master file A detailed description of the pharmacovigilance system used by the marketing authorisation holder with respect to one or more authorised medicinal products. The national competent authority may at any time ask the marketing authorisation holder to submit a copy of the pharmacovigilance system master file. The marketing authorisation holder shall submit the copy at the latest 7 days after receipt of the request. 37
38 DDPS- PhVS master file The new Directive and RSegulation do not contain details on the content and maintenance of the PhV-MF. Elements of its content and maintenance will be required by implementing measures under consultation and the Good Vigilance Practices guidance under development. Important changes: No content of the PhVS-MF is submitted as part of the MAA Review of the PhVS-MF is on request 38
39 DDPS- PhVS master file D 104 QPPV : should reside and carry out tasks in the EU Legal basis for MSs to request a national nominated person for pharmacovigilance is introduced National nominated persons should report to the QPPV 39
40 DDPS- PhVS master file Currently DDPS Variations, type I or II Changes to PSMF content will not require variations Variations are only required (Directive article 8 ) change in QPPV PhVS-MF location The current proposal for the implementing measures: Specify changes to PhVSMF location notified via product list (Article 57 of the Regulation ) this address is the same as that of the QPPV. Since MAHs will simply be notifying NCAs/ EMA of an administrative change, only type IA variation are proposed. 40
41 Overzicht presentatie Introductie,doel van de veranderingen Wetgeving Verordening Richtlijn Implementatie maatregelen Belangrijke veranderingen in vogelvlucht PRAC PASS PhVS MF Additional monitoring 41
42 Additional monitoring Regulation art 23/Directive art 11 The Agency shall, in collaboration with the Member States, set up, maintain and make public a list of medicinal products that are subject to additional monitoring. That list shall include the names and active substances of: Medicinal products authorised in the Union that contain a new active substance which, on 1 January 2011, was not contained in any medicinal product authorised in the Union; any biological medicinal produc t not covered by point (a) authorised after 1 January At the request of the Commission, following consultation with the PRAC (Regulation, CAPs) At the request of the National Competent Authority following consultation with the PRAC (Directive, non CAPs) 42
43 Additional monitoring The list shall include an electronic link to the product information the summary of the risk management plan. The Agency shall remove a product from the list: 5 years after the Union Reference date (art 107) Unless 43
44 Additional monitoring R23/D59 SPC and PL shall include the statement This medicinal product is subject to additional monitoring. Statement preceded by a black symbol (selected by the Commission following a recommendation of the PRAC) followed by an appropriate standardised explanatory sentence For all medicinal products, a standardised text shall be included, expressly asking patients to communicate any suspected adverse reaction to his/her doctor, pharmacist, healthcare professional or directly to the national spontaneous reporting system referred to in Article 107a(1), specifying the different ways of reporting available (electronic reporting, postal address and/or others) in compliance with the second subparagraph of Article 107a(1) 44
45 EU the United States of Europe EU legislation: 27 different sets of national legislation 45
46 Volume 9A wordt : GVP Modulair van opzet Ontwikkeld door EMA samen met lidstaten Uit voor pubieke consultatie ( 8 weken) Planning: gefaseerd eerste wave januari 2012
47 GUIDANCE ON GOOD PHARMACOVIGILANCE PRACTICES INTRODUCTION Legal Basis and Structure of Pharmacovigilance Guidance MODULE I Pharmacovigilance Systems and their Quality Systems MODULE II Pharmacovigilance System Master File MODULE III Pharmacovigilance Inspections MODULE IV Audits MODULE V Risk Management Systems MODULE VI Data Management of Individual Case Safety Reports MODULE VII Periodic Safety Update Reports MODULE VIII Post-Authorisation Safety Studies MODULE IX Post-Authorisation Efficacy Studies (to be decided later) MODULE X Detection and Management of Signals and Information MODULE XI Additional Monitoring MODULE XII Public Participation in Pharmacovigilance MODULE XIII Continuous Pharmacovigilance, Ongoing Benefit-Risk Evaluation, Regulatory Action and Planning of Public Communication MODULE XIV Incident Management MODULE XV Referral Procedures for Safety Reasons MODULE XVI Safety Communications MODULE XVII Tools, Educational Materials and Effectiveness Measurement for Risk Minimisation 47
48 Thank you! Questions? Better protecting the patients Be aware of the system and your role
49 49
Detailed guidance AEs reporting in clinical trials ( CT-3 ) Stefano Bonato, M.D. Pharmacovigilance Country Head Bayer HealthCare Pharmaceuticals
 Stefano Bonato, M.D. Pharmacovigilance Country Head Bayer HealthCare Pharmaceuticals") Detailed guidance AEs reporting in clinical trials ( CT-3 ) Stefano Bonato, M.D. Pharmacovigilance Country Head Bayer HealthCare Pharmaceuticals Pharmacovigilance costs 0.12%-0.22% of hospital admissions
Detailed guidance AEs reporting in clinical trials ( CT-3 ) Stefano Bonato, M.D. Pharmacovigilance Country Head Bayer HealthCare Pharmaceuticals Pharmacovigilance costs 0.12%-0.22% of hospital admissions
Agence fédérale des médicaments et des produits de santé
 Agence fédérale des médicaments et des produits de santé From Risk Management Plan to Risk Minimization Activities New Royal Decree: Status and General Explanation Role of the Pharmacovigilance Department
Agence fédérale des médicaments et des produits de santé From Risk Management Plan to Risk Minimization Activities New Royal Decree: Status and General Explanation Role of the Pharmacovigilance Department
Guideline on good pharmacovigilance practices (GVP)
") 1 2 20 February 2012 EMA/541760/2011 3 4 Guideline on good pharmacovigilance practices (GVP) Module I Pharmacovigilance systems and their quality systems Draft finalised by the Agency in collaboration
1 2 20 February 2012 EMA/541760/2011 3 4 Guideline on good pharmacovigilance practices (GVP) Module I Pharmacovigilance systems and their quality systems Draft finalised by the Agency in collaboration
Guideline on good pharmacovigilance practices (GVP)
") 22 June 2012 EMA/541760/2011 Guideline on good pharmacovigilance practices (GVP) Module I Pharmacovigilance systems and their quality systems Draft finalised by the Agency in collaboration with Member
22 June 2012 EMA/541760/2011 Guideline on good pharmacovigilance practices (GVP) Module I Pharmacovigilance systems and their quality systems Draft finalised by the Agency in collaboration with Member
Questions & answers on signal management
 23 October 2015 EMA/261758/2013 Inspections and Human Medicines Pharmacovigilance Division This document addresses a number of questions which stakeholders, in particular marketing authorisation holders
23 October 2015 EMA/261758/2013 Inspections and Human Medicines Pharmacovigilance Division This document addresses a number of questions which stakeholders, in particular marketing authorisation holders
Post-authorisation safety studies and the EU PAS Register
 Post-authorisation safety studies and the EU PAS Register Xavier Kurz European Medicines Agency EMA, 11 October 2012 An agency of the European Union Content of the presentation 1. PASS: definition and
Post-authorisation safety studies and the EU PAS Register Xavier Kurz European Medicines Agency EMA, 11 October 2012 An agency of the European Union Content of the presentation 1. PASS: definition and
Guideline on good pharmacovigilance practices (GVP)
") 9 April 2013 EMA/816573/2011 Rev 1* Guideline on good pharmacovigilance practices (GVP) Module II Pharmacovigilance system master file (Rev 1) Draft of first version finalised by the Agency in collaboration
9 April 2013 EMA/816573/2011 Rev 1* Guideline on good pharmacovigilance practices (GVP) Module II Pharmacovigilance system master file (Rev 1) Draft of first version finalised by the Agency in collaboration
Guideline on good pharmacovigilance practices (GVP)
") 19 April 2013 EMA/813938/2011 Rev 1* Guideline on good pharmacovigilance practices (GVP) Module VIII Post-authorisation safety studies (Rev 1) Draft of first version finalised by the Agency in collaboration
19 April 2013 EMA/813938/2011 Rev 1* Guideline on good pharmacovigilance practices (GVP) Module VIII Post-authorisation safety studies (Rev 1) Draft of first version finalised by the Agency in collaboration
One-year report on human medicines pharmacovigilance tasks of the European Medicines Agency
 8 April 2014 EMA/171322/2014 Pharmacovigilance Department One-year report on human medicines pharmacovigilance tasks of the European Medicines Agency Reporting period: 2 July 2012 to 1 July 2013 7 Westferry
8 April 2014 EMA/171322/2014 Pharmacovigilance Department One-year report on human medicines pharmacovigilance tasks of the European Medicines Agency Reporting period: 2 July 2012 to 1 July 2013 7 Westferry
Pharmacovigilance System Master File (PSMF), QPPV and audits
, QPPV and audits") Federal Agency for Medicines and Health Products (FAMHP) Pharmacovigilance System Master File (PSMF), QPPV and audits Matthijs Nele, PhV Inspector, DG Inspection BRAS - FAMHP/NM/ 1 Content Introduction
Federal Agency for Medicines and Health Products (FAMHP) Pharmacovigilance System Master File (PSMF), QPPV and audits Matthijs Nele, PhV Inspector, DG Inspection BRAS - FAMHP/NM/ 1 Content Introduction
Guidance notes on collection of drug safety information by employees and agents of pharmaceutical companies
 Guidance notes on collection of drug safety information by employees and agents of pharmaceutical companies March 2013 Guidance notes on collection of drug safety information by employees and agents of
Guidance notes on collection of drug safety information by employees and agents of pharmaceutical companies March 2013 Guidance notes on collection of drug safety information by employees and agents of
ICRIN Seminar on EU Regulation of Clinical Trials
 ICRIN Seminar on EU Regulation of Clinical Trials 12 th March 2013, Dublin J. Michael Morris Director Scientific Affairs IRISH MEDICINES BOARD 28/03/2013 Slide 1 Overview Clinical Trial (CT) legislation
ICRIN Seminar on EU Regulation of Clinical Trials 12 th March 2013, Dublin J. Michael Morris Director Scientific Affairs IRISH MEDICINES BOARD 28/03/2013 Slide 1 Overview Clinical Trial (CT) legislation
Guideline on good pharmacovigilance practices (GVP)
") 22 June 2012 EMA/827661/2011 Guideline on good pharmacovigilance practices (GVP) Module IX Signal management Draft finalised by the Agency in collaboration with Member States and submitted to ERMS FG 19
22 June 2012 EMA/827661/2011 Guideline on good pharmacovigilance practices (GVP) Module IX Signal management Draft finalised by the Agency in collaboration with Member States and submitted to ERMS FG 19
Competentes en Medicamentos
 VIII Encuentro de Autoridades Competentes en Medicamentos de los Países Iberoamericanos (EAMI) 12-14 de Mayo 2010 Madrid MARTIN TERBERGER Jefe de la Unidad de Productos Farmacéuticos Nuevas iniciativas
VIII Encuentro de Autoridades Competentes en Medicamentos de los Países Iberoamericanos (EAMI) 12-14 de Mayo 2010 Madrid MARTIN TERBERGER Jefe de la Unidad de Productos Farmacéuticos Nuevas iniciativas
Report to the European Commission on Pharmacovigilance audits carried out in the Medicines Evaluation Board, The Netherlands period of time from
 Report to the European Commission on Pharmacovigilance audits carried out in the Medicines Evaluation Board, The Netherlands period of time from September 2013 to September 2015 1. INTRODUCTION Applicable
Report to the European Commission on Pharmacovigilance audits carried out in the Medicines Evaluation Board, The Netherlands period of time from September 2013 to September 2015 1. INTRODUCTION Applicable
How To Understand The Paediatric Regulation
 Paediatric Use Marketing Authorisation (PUMA) from a National Competent Authority Point of View DGRA Annual Congress 2006 Bonn, 10. May 2006 Dr. Susanne Keitel Content PUMA The Future Background Legislative
Paediatric Use Marketing Authorisation (PUMA) from a National Competent Authority Point of View DGRA Annual Congress 2006 Bonn, 10. May 2006 Dr. Susanne Keitel Content PUMA The Future Background Legislative
Seminario SSFA. New Guideline on Good Pharmacovigilance Practice (GVP) Fare clic per modificare lo stile del sottotitolo dello schema
 Fare clic per modificare lo stile del sottotitolo dello schema") Seminario SSFA New Guideline on Good Pharmacovigilance Practice (GVP) Pharmacovigilance Systems and their Quality Systems Milano, 20th April 2012 GIULIA M. VALSECCHI The proposed GVP - Module 1 Guidance
Seminario SSFA New Guideline on Good Pharmacovigilance Practice (GVP) Pharmacovigilance Systems and their Quality Systems Milano, 20th April 2012 GIULIA M. VALSECCHI The proposed GVP - Module 1 Guidance
Regulatory approval routes in the European System for Medicinal Products
 Regulatory approval routes in the European System for Medicinal Products Cardiovascular Combination Pharmacotherapy Global Summit, Melbourne, 8 th May 2014 Presented by: Kevin Blake Human Medicines Research
Regulatory approval routes in the European System for Medicinal Products Cardiovascular Combination Pharmacotherapy Global Summit, Melbourne, 8 th May 2014 Presented by: Kevin Blake Human Medicines Research
BEST PRACTICE GUIDE for Handling of Periodic Safety Update Reports
 EMEA/CMDv/408477/2007 12 March 2009 BEST PRACTICE GUIDE for Handling of Periodic Safety Update Reports Edition number : 00 Edition date: 12 March 2009 Implementation date : 27 March 2009 CMD(v) Secretariat:
EMEA/CMDv/408477/2007 12 March 2009 BEST PRACTICE GUIDE for Handling of Periodic Safety Update Reports Edition number : 00 Edition date: 12 March 2009 Implementation date : 27 March 2009 CMD(v) Secretariat:
EMA pharmacovigilance system manual
 30 June 2014 EMA/623550/2013 Inspections and Human Medicines Pharmacovigilance Division Version 1.1 30 Churchill Place Canary Wharf London E14 5EU United Kingdom Telephone +44 (0)20 3660 6000 Facsimile
30 June 2014 EMA/623550/2013 Inspections and Human Medicines Pharmacovigilance Division Version 1.1 30 Churchill Place Canary Wharf London E14 5EU United Kingdom Telephone +44 (0)20 3660 6000 Facsimile
Guideline on good pharmacovigilance practices (GVP)
") 1 2 20 February 2012 EMA/876333/2011 3 4 Guideline on good pharmacovigilance practices (GVP) Annex I - Definitions Draft finalised by the Agency in collaboration with Member States 7 February 2012 Definitions
1 2 20 February 2012 EMA/876333/2011 3 4 Guideline on good pharmacovigilance practices (GVP) Annex I - Definitions Draft finalised by the Agency in collaboration with Member States 7 February 2012 Definitions
Comparison of EU-Pharmacovigilance System Master File (PSMF) with US System
 with US System") Comparison of EU-Pharmacovigilance System Master File (PSMF) with US System Wissenschaftliche Prüfungsarbeit Zur Eralngung des Titels Master of Drug Regulatory Affairs der Mathematisch-Naturwissenschaftlichen
Comparison of EU-Pharmacovigilance System Master File (PSMF) with US System Wissenschaftliche Prüfungsarbeit Zur Eralngung des Titels Master of Drug Regulatory Affairs der Mathematisch-Naturwissenschaftlichen
Opinion/ Commission. Notification. Decision. Issued 2 / affected 3 amended on. 30/07/2015 Annex II, Labelling and PL
 Procedural steps taken and scientific information after the authorisation Application Scope Opinion/ Commission Product Summary number Notification Decision Information 1 issued on Issued 2 / affected
Procedural steps taken and scientific information after the authorisation Application Scope Opinion/ Commission Product Summary number Notification Decision Information 1 issued on Issued 2 / affected
COMMISSION STAFF WORKING DOCUMENT EXECUTIVE SUMMARY OF THE IMPACT ASSESSMENT. Accompanying the document
 EUROPEAN COMMISSION Brussels, 26.6.2013 SWD(2013) 235 final COMMISSION STAFF WORKING DOCUMENT EXECUTIVE SUMMARY OF THE IMPACT ASSESSMENT Accompanying the document Proposal for a Regulation of the European
EUROPEAN COMMISSION Brussels, 26.6.2013 SWD(2013) 235 final COMMISSION STAFF WORKING DOCUMENT EXECUTIVE SUMMARY OF THE IMPACT ASSESSMENT Accompanying the document Proposal for a Regulation of the European
EUROPEAN MEDICINES PRACTICES AND TOOLS AGENCY DRUG SAFETY
 EUROPEAN MEDICINES AGENCY DRUG SAFETY PRACTICES AND TOOLS Henry Fitt, Kevin Blake, Xavier Kurz Pharmacovigilance & Risk Management European Medicines Agency, London Contents Eudravigilance & Signal Detection
EUROPEAN MEDICINES AGENCY DRUG SAFETY PRACTICES AND TOOLS Henry Fitt, Kevin Blake, Xavier Kurz Pharmacovigilance & Risk Management European Medicines Agency, London Contents Eudravigilance & Signal Detection
Report from the CMDh meeting held on 21-23 September 2015. !!! 3 months to go until the mandatory use of the electronic application form!!!
 Report from the meeting held on 21-23 September 2015!!! 3 months to go until the mandatory use of the electronic application form!!! Pharmacovigilance positions following PSUSA procedure for only nationally
Report from the meeting held on 21-23 September 2015!!! 3 months to go until the mandatory use of the electronic application form!!! Pharmacovigilance positions following PSUSA procedure for only nationally
Data submission of authorised medicines in the European Union
 23 February 2015 EMA/471367/2014, Rev. 1 1 Business Data and Support Department Data submission of authorised medicines in the European Union Outlines on Article 57(2) of Regulation (EC) No 726/2004 1
23 February 2015 EMA/471367/2014, Rev. 1 1 Business Data and Support Department Data submission of authorised medicines in the European Union Outlines on Article 57(2) of Regulation (EC) No 726/2004 1
Guidance for the format and content of the protocol of non-interventional post-authorisation safety studies
 26 September 2012 EMA/623947/2012 Patient Health Protection Guidance for the format and content of the protocol of non-interventional post-authorisation Introduction From 10 January 2013, marketing authorisation
26 September 2012 EMA/623947/2012 Patient Health Protection Guidance for the format and content of the protocol of non-interventional post-authorisation Introduction From 10 January 2013, marketing authorisation
Good practice guide on recording, coding, reporting and assessment of medication errors
 1 2 3 14 April 2015 EMA/762563/2014 Pharmacovigilance Risk Assessment Committee (PRAC) 4 5 6 Good practice guide on recording, coding, reporting and assessment of medication errors Draft Draft finalised
1 2 3 14 April 2015 EMA/762563/2014 Pharmacovigilance Risk Assessment Committee (PRAC) 4 5 6 Good practice guide on recording, coding, reporting and assessment of medication errors Draft Draft finalised
EU PAS Register Guide
 29 March 2016 EMA/613603/2012 (currently ENCePP E-Register of Studies) The EU PAS Register is temporarily hosted on the ENCePP website www.encepp.eu 1. Introduction... 2 2. ENCePP E-Register as a temporary
29 March 2016 EMA/613603/2012 (currently ENCePP E-Register of Studies) The EU PAS Register is temporarily hosted on the ENCePP website www.encepp.eu 1. Introduction... 2 2. ENCePP E-Register as a temporary
VOLUME 2A Procedures for marketing authorisation CHAPTER 1 MARKETING AUTHORISATION. November 2005
 EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Consumer goods Pharmaceuticals Brussels, ENTR/F2/BL D(2002) NOTICE TO APPLICANTS Revision 3 VOLUME 2A Procedures for marketing authorisation CHAPTER 1
EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Consumer goods Pharmaceuticals Brussels, ENTR/F2/BL D(2002) NOTICE TO APPLICANTS Revision 3 VOLUME 2A Procedures for marketing authorisation CHAPTER 1
COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS GUIDELINE FOR THE CONDUCT OF POST-MARKETING SURVEILLANCE STUDIES OF VETERINARY MEDICINAL PRODUCTS
 The European Agency for the Evaluation of Medicinal Products Veterinary Medicines and Information Technology EMEA/CVMP/044/99-FINAL COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS GUIDELINE FOR THE CONDUCT
The European Agency for the Evaluation of Medicinal Products Veterinary Medicines and Information Technology EMEA/CVMP/044/99-FINAL COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS GUIDELINE FOR THE CONDUCT
The EMA current activities towards ISO IDMP implementation
 The EMA current activities towards ISO IDMP implementation An overview Ginas meeting 11 June 2014 Presented by: Ilaria Del Seppia, B-BD-DCM EMA An agency of the European Union Outlines EMA Vision: Objectives
The EMA current activities towards ISO IDMP implementation An overview Ginas meeting 11 June 2014 Presented by: Ilaria Del Seppia, B-BD-DCM EMA An agency of the European Union Outlines EMA Vision: Objectives
Stakeholder Meeting, 7 th June 2013 An inspector s perspective considerations for patient support and reimbursement programmes
 Stakeholder Meeting, 7 th June 2013 An inspector s perspective considerations for patient support and reimbursement programmes Dr Anya Sookoo, Expert Inspector, Inspections, Enforcement & Standards Division,
Stakeholder Meeting, 7 th June 2013 An inspector s perspective considerations for patient support and reimbursement programmes Dr Anya Sookoo, Expert Inspector, Inspections, Enforcement & Standards Division,
Proposal for a RECOMMENDATION OF THE EUROPEAN COMMISSION
 EUROPEAN COMMISSION Brussels, XXX [ ] (2013) XXX draft Proposal for a RECOMMENDATION OF THE EUROPEAN COMMISSION Providing minimum principles for the exploration and production of hydrocarbons (especially
EUROPEAN COMMISSION Brussels, XXX [ ] (2013) XXX draft Proposal for a RECOMMENDATION OF THE EUROPEAN COMMISSION Providing minimum principles for the exploration and production of hydrocarbons (especially
REGULATION (EEC) No 2309/93
 No 2309/93") REGULATION (EEC) No 2309/93 Council Regulation (EEC) No 2309/93 of 22 July 1993 laying down Community procedures for the authorization and supervision of medicinal products for human and veterinary use
REGULATION (EEC) No 2309/93 Council Regulation (EEC) No 2309/93 of 22 July 1993 laying down Community procedures for the authorization and supervision of medicinal products for human and veterinary use
EudraVigilance stakeholder change management plan
 26 October 2015 EMA/797114/2014 Information Management Division Consultation of Project Maintenance Group 1 15 July 2015 Consultation of Eudravigilance Expert Working Group 23 September 2015 Consultation
26 October 2015 EMA/797114/2014 Information Management Division Consultation of Project Maintenance Group 1 15 July 2015 Consultation of Eudravigilance Expert Working Group 23 September 2015 Consultation
The Clinical Trials Regulation EU No 536/2014: and Phase I trials
 The Clinical Trials Regulation EU No 536/2014: and Phase I trials EUFEMED, Brussels, 20 May 2015 Presented by Fergus Sweeney Head, Inspections and Human Medicines Pharmacovigilance An agency of the European
The Clinical Trials Regulation EU No 536/2014: and Phase I trials EUFEMED, Brussels, 20 May 2015 Presented by Fergus Sweeney Head, Inspections and Human Medicines Pharmacovigilance An agency of the European
RAPID ALERT SYSTEM (RAS) IN PHARMACOVIGILANCE
 IN PHARMACOVIGILANCE") 3BC6a RAPID ALERT SYSTEM (RAS) IN PHARMACOVIGILANCE Guideline Title Rapid Alert System (RAS) in Pharmacovigilance Legislative basis Directive 65/65/EEC as amended, Council Regulation 2309/93 Date of first
3BC6a RAPID ALERT SYSTEM (RAS) IN PHARMACOVIGILANCE Guideline Title Rapid Alert System (RAS) in Pharmacovigilance Legislative basis Directive 65/65/EEC as amended, Council Regulation 2309/93 Date of first
EphMRA Adverse Event Reporting Guidelines 2015
 EphMRA Adverse Event Reporting Guidelines 2015 Based upon the Guideline on good pharmacovigilance practices (GVP) Module VI Management and reporting of adverse reactions to medicinal products European
EphMRA Adverse Event Reporting Guidelines 2015 Based upon the Guideline on good pharmacovigilance practices (GVP) Module VI Management and reporting of adverse reactions to medicinal products European
EphMRA Adverse Event Reporting Guidelines 2013
 EphMRA Adverse Event Reporting Guidelines 2013 Based upon the Guideline on good pharmacovigilance practices (GVP) Module VI Management and reporting of adverse reactions to medicinal products European
EphMRA Adverse Event Reporting Guidelines 2013 Based upon the Guideline on good pharmacovigilance practices (GVP) Module VI Management and reporting of adverse reactions to medicinal products European
Good practice guide on recording, coding, reporting and assessment of medication errors
 23 October 2015 EMA/762563/2014 Pharmacovigilance Risk Assessment Committee (PRAC) Good practice guide on recording, coding, reporting and assessment of medication errors Final Draft finalised by Project
23 October 2015 EMA/762563/2014 Pharmacovigilance Risk Assessment Committee (PRAC) Good practice guide on recording, coding, reporting and assessment of medication errors Final Draft finalised by Project
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP)
") European Medicines Agency Evaluation of Medicines for Human Use London, 19 July 2007 Doc. Ref: EMEA/27170/2006 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON COMPASSIONATE USE OF MEDICINAL
European Medicines Agency Evaluation of Medicines for Human Use London, 19 July 2007 Doc. Ref: EMEA/27170/2006 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) GUIDELINE ON COMPASSIONATE USE OF MEDICINAL
EMA Update Clinical Trials
 EMA Update Clinical Trials Fergus Sweeney, Head, Compliance and Inspections, European Medicines Agency 16 October 2012 An agency of the European Union Disclaimer The views presented in this presentation/these
EMA Update Clinical Trials Fergus Sweeney, Head, Compliance and Inspections, European Medicines Agency 16 October 2012 An agency of the European Union Disclaimer The views presented in this presentation/these
Clinical research: where are we with the new (Paediatric) RC trial Regulation
 RC trial Regulation") where are we with the new (Paediatric) RC trial Regulation, MD, PhD Ethical Committee DEEP Former member of the PDCO EMA With the aid of Fabio D'Atri European commission and Anabela Marcal of EMA The new
where are we with the new (Paediatric) RC trial Regulation, MD, PhD Ethical Committee DEEP Former member of the PDCO EMA With the aid of Fabio D'Atri European commission and Anabela Marcal of EMA The new
Response of the German Medical Association
 Response of the German Medical Association to the European Commission proposal for a regulation of the European Parliament and of the Council on clinical trials on medicinal products for human use, and
Response of the German Medical Association to the European Commission proposal for a regulation of the European Parliament and of the Council on clinical trials on medicinal products for human use, and
Clinical trials regulation
 Clinical trials regulation The Proposal for a Regulation of the European Parliament and of the Council on Clinical Trials on Medicinal Products for Human Use and Repealing Directive 2001/20/EC an update
Clinical trials regulation The Proposal for a Regulation of the European Parliament and of the Council on Clinical Trials on Medicinal Products for Human Use and Repealing Directive 2001/20/EC an update
European Regulatory Newsletter July - September 2013
 European Regulatory Newsletter July - September 2013 Introduction CROMSOURCE is committed to sharing our expertise with our clients and future clients. This reflects the first part of our Advise Agree
European Regulatory Newsletter July - September 2013 Introduction CROMSOURCE is committed to sharing our expertise with our clients and future clients. This reflects the first part of our Advise Agree
Working Party on Control of Medicines and Inspections. Final Version of Annex 16 to the EU Guide to Good Manufacturing Practice
 Version 8 (final) EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Single market, regulatory environment, industries under vertical legislation Pharmaceuticals and cosmetics Brussels, July 2001 S\common\legal-legislation\75-319nd81-851\91-356\eudralexvol4\Annex
Version 8 (final) EUROPEAN COMMISSION ENTERPRISE DIRECTORATE-GENERAL Single market, regulatory environment, industries under vertical legislation Pharmaceuticals and cosmetics Brussels, July 2001 S\common\legal-legislation\75-319nd81-851\91-356\eudralexvol4\Annex
Reflection Paper on Medicinal Product Supply Shortages Caused by Manufacturing/GMP Compliance Problems
 14 September 2012 /590745/2012 Patient Health Protection Reflection Paper on Medicinal Product Supply Shortages Caused by Manufacturing/GMP 1. Introduction Ensuring the security and Good Manufacturing
14 September 2012 /590745/2012 Patient Health Protection Reflection Paper on Medicinal Product Supply Shortages Caused by Manufacturing/GMP 1. Introduction Ensuring the security and Good Manufacturing
Implementation strategy for ISO IDMP in EU
 Implementation strategy for ISO IDMP in EU Paolo Alcini Head of Data Standardisation and Analytics Business Data & Analytics Information Management EMA An agency of the European Union The EU ISO IDMP Task
Implementation strategy for ISO IDMP in EU Paolo Alcini Head of Data Standardisation and Analytics Business Data & Analytics Information Management EMA An agency of the European Union The EU ISO IDMP Task
Clinical trials: from European perspective to National implementation. CTFG / FAMHP / pharma.be. Brussels, 19 November 2010
 Clinical trials: from European perspective to National implementation CTFG / FAMHP / pharma.be Brussels, 19 November 2010 Safety in clinical trials: From detection to decision How safety events are captured
Clinical trials: from European perspective to National implementation CTFG / FAMHP / pharma.be Brussels, 19 November 2010 Safety in clinical trials: From detection to decision How safety events are captured
Risk Management Plan (RMP) Guidance (Draft)
 Guidance (Draft)") Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare Translated by Pharmaceuticals and Medical Devices Agency Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour
Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour and Welfare Translated by Pharmaceuticals and Medical Devices Agency Pharmaceutical and Food Safety Bureau, Ministry of Health, Labour
PHV- 4 version 1 ELECTRONIC ADVERSE DRUG REACTION REPORTING
 PHV- 4 version 1 ELECTRONIC ADVERSE DRUG REACTION REPORTING This guideline supersedes guideline PHV 4 as of September 16 2008. 1. Introduction and general provisions 1.1 Purpose of the guideline The guideline
PHV- 4 version 1 ELECTRONIC ADVERSE DRUG REACTION REPORTING This guideline supersedes guideline PHV 4 as of September 16 2008. 1. Introduction and general provisions 1.1 Purpose of the guideline The guideline
EU DIRECTIVE ON GOOD CLINICAL PRACTICE IN CLINICAL TRIALS DH & MHRA BRIEFING NOTE
 EU DIRECTIVE ON GOOD CLINICAL PRACTICE IN CLINICAL TRIALS DH & MHRA BRIEFING NOTE Purpose 1. The Clinical Trials Directive 2001/20/EC heralds certain additional responsibilities for the Medicines and Healthcare
EU DIRECTIVE ON GOOD CLINICAL PRACTICE IN CLINICAL TRIALS DH & MHRA BRIEFING NOTE Purpose 1. The Clinical Trials Directive 2001/20/EC heralds certain additional responsibilities for the Medicines and Healthcare
Monitoring of medical literature and the entry of relevant information into the EudraVigilance database by the European Medicines Agency
 28 August 2015 EMA/386784/2015 Inspections and Human Medicines Pharmacovigilance Division Monitoring of medical literature and the entry of relevant information into the EudraVigilance database by the
28 August 2015 EMA/386784/2015 Inspections and Human Medicines Pharmacovigilance Division Monitoring of medical literature and the entry of relevant information into the EudraVigilance database by the
Risk Management Plan for Drug Establishments
 Republic of the Philippines Department of Health FOOD AND DRUG ADMINISTRATION Risk Management Plan for Drug Establishments Center for Drug Regulation and Research Food and Drug Administration 17 December
Republic of the Philippines Department of Health FOOD AND DRUG ADMINISTRATION Risk Management Plan for Drug Establishments Center for Drug Regulation and Research Food and Drug Administration 17 December
COMMISSION REGULATION (EU) No /.. of XXX
 No /.. of XXX") EUROPEAN COMMISSION Brussels, XXX D... [ ](2012) XXX draft COMMISSION REGULATION (EU) No /.. of XXX establishing a Union Registry pursuant to Directive 2003/87/EC of the European Parliament and of the
EUROPEAN COMMISSION Brussels, XXX D... [ ](2012) XXX draft COMMISSION REGULATION (EU) No /.. of XXX establishing a Union Registry pursuant to Directive 2003/87/EC of the European Parliament and of the
MedDRA in pharmacovigilance industry perspective
 MedDRA in pharmacovigilance industry SFDA ICH MedDRA Workshop, Beijing, 13 14 May 2011 Christina Winter, MD, FFPM Medical director GlaxoSmithKline R&D / ICH MedDRA Management Board 2011 ICH International
MedDRA in pharmacovigilance industry SFDA ICH MedDRA Workshop, Beijing, 13 14 May 2011 Christina Winter, MD, FFPM Medical director GlaxoSmithKline R&D / ICH MedDRA Management Board 2011 ICH International
Emergence of Compassionate Use programmes
 Emergence of Compassionate Use programmes Rosanna Melchior, PharmD, MS IDRAC, Thomson Scientific Overview What is compassionate use (CU) Overview of CU in Europe Authorities and Sponsors Roles and Responsibilities
Emergence of Compassionate Use programmes Rosanna Melchior, PharmD, MS IDRAC, Thomson Scientific Overview What is compassionate use (CU) Overview of CU in Europe Authorities and Sponsors Roles and Responsibilities
Marketing Authorisation: The Evaluation Process
 1 st EMEA Workshop for Micro, Small and Medium- Sized Enterprises (SMEs) Navigating the Regulatory Maze Marketing Authorisation: The Evaluation Process Dr Evdokia Korakianiti Scientific Administrator Centralised
1 st EMEA Workshop for Micro, Small and Medium- Sized Enterprises (SMEs) Navigating the Regulatory Maze Marketing Authorisation: The Evaluation Process Dr Evdokia Korakianiti Scientific Administrator Centralised
The European Union regulatory network incident management plan for medicines for human use
 28 July 2015 Inspection and Human Medicines Pharmacovigilance The European Union regulatory network incident management plan for medicines for human use 1. Introduction The continuous monitoring of medicinal
28 July 2015 Inspection and Human Medicines Pharmacovigilance The European Union regulatory network incident management plan for medicines for human use 1. Introduction The continuous monitoring of medicinal
EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL
 Ref. Ares(2011)1044649-03/10/2011 EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Health systems and products Pharmaceuticals SANCO/D3/RSR/iv(2011)ddg1.d3. 1137738 Update 1 October 2011 HANDLING
Ref. Ares(2011)1044649-03/10/2011 EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Health systems and products Pharmaceuticals SANCO/D3/RSR/iv(2011)ddg1.d3. 1137738 Update 1 October 2011 HANDLING
The New EU Clinical Trial Regulation Potential Impacts on Sites
 The New EU Clinical Trial Regulation Potential Impacts on Sites Angela Papa Associate Director, Clinical Management PPD Pierre-Frédéric Omnes Director, Site Start-Up and Regulatory INC Research Faculty
The New EU Clinical Trial Regulation Potential Impacts on Sites Angela Papa Associate Director, Clinical Management PPD Pierre-Frédéric Omnes Director, Site Start-Up and Regulatory INC Research Faculty
Quick Response (QR) codes in the labelling and package leaflet of centrally authorised 1 medicinal products
 codes in the labelling and package leaflet of centrally authorised 1 medicinal products") 22 July 2015 EMA/493897/2015 Human Medicines Evaluation Division Quick Response (QR) codes in the labelling and package leaflet of centrally authorised 1 medicinal General principles of acceptability and
22 July 2015 EMA/493897/2015 Human Medicines Evaluation Division Quick Response (QR) codes in the labelling and package leaflet of centrally authorised 1 medicinal General principles of acceptability and
Opinion/ Commission. Notification 1. Decision. Information issued on. Issued 2 / affected 3 amended on
 Procedural steps taken and scientific information after the authorisation Application Scope Opinion/ Commission Product Summary number Notification 1 Decision Information issued on Issued 2 / affected
Procedural steps taken and scientific information after the authorisation Application Scope Opinion/ Commission Product Summary number Notification 1 Decision Information issued on Issued 2 / affected
Guidelines. of 16.05.2013
 EUROPEAN COMMISSION Brussels, 16.05.2013 C (2013) 2804 Guidelines of 16.05.2013 on the details of the various categories of variations, on the operation of the procedures laid down in Chapters, a, I and
EUROPEAN COMMISSION Brussels, 16.05.2013 C (2013) 2804 Guidelines of 16.05.2013 on the details of the various categories of variations, on the operation of the procedures laid down in Chapters, a, I and
COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS (CVMP)
 COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS (CVMP)") European Medicines Agency Veterinary Medicines and Inspections London, 15 April 2005 EMEA/CVMP/134/02 Rev 1 CPMP/QWP/227/02 Rev 1 COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) COMMITTEE FOR VETERINARY
European Medicines Agency Veterinary Medicines and Inspections London, 15 April 2005 EMEA/CVMP/134/02 Rev 1 CPMP/QWP/227/02 Rev 1 COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS (CPMP) COMMITTEE FOR VETERINARY
Official Journal of the European Union. (Acts whose publication is obligatory)
") 27.12.2006 L 378/1 I (Acts whose publication is obligatory) REGULATION (EC) No 1901/2006 OF THE EUROPEAN PARLIAMT AND OF THE COUNCIL of 12 December 2006 on medicinal products for paediatric use and amending
27.12.2006 L 378/1 I (Acts whose publication is obligatory) REGULATION (EC) No 1901/2006 OF THE EUROPEAN PARLIAMT AND OF THE COUNCIL of 12 December 2006 on medicinal products for paediatric use and amending
Guideline on good pharmacovigilance practices (GVP)
") 15 April 2014 EMA/838713/2011 Rev 1* Guideline on good pharmacovigilance practices (GVP) Module V Risk management systems (Rev 1) Draft finalised by the Agency in collaboration with Member States and submitted
15 April 2014 EMA/838713/2011 Rev 1* Guideline on good pharmacovigilance practices (GVP) Module V Risk management systems (Rev 1) Draft finalised by the Agency in collaboration with Member States and submitted
ISSUING THE AIR OPERATOR CERTIFICATE, OPERATIONS SPECIFICATIONS, AND COMPLETING THE CERTIFICATION REPORT
 ISSUING THE AIR OPERATOR CERTIFICATE, OPERATIONS SPECIFICATIONS, AND COMPLETING THE CERTIFICATION REPORT GUIDANCE MATERIAL FOR INSPECTORS CA AOC-017 AIR OPERATOR CERTIFICATION RECORD OF AMENDMENTS Amendment
ISSUING THE AIR OPERATOR CERTIFICATE, OPERATIONS SPECIFICATIONS, AND COMPLETING THE CERTIFICATION REPORT GUIDANCE MATERIAL FOR INSPECTORS CA AOC-017 AIR OPERATOR CERTIFICATION RECORD OF AMENDMENTS Amendment
Electronic exchanges of individual case safety reports (ICSRs) with ANSM (National Agency of Medicine and Health Product safety)
 with ANSM (National Agency of Medicine and Health Product safety)") Pharmacovigilance information for pharmaceutical companies Electronic exchanges of individual case safety reports (ICSRs) with ANSM (National Agency of Medicine and Health Product safety) This document
Pharmacovigilance information for pharmaceutical companies Electronic exchanges of individual case safety reports (ICSRs) with ANSM (National Agency of Medicine and Health Product safety) This document
Guidance notes on the management of adverse events and product complaints from digital media
 Guidance notes on the management of adverse events and product complaints from digital media Guidance notes on the management of adverse events and product complaints from digital media ABPI Pharmacovigilance
Guidance notes on the management of adverse events and product complaints from digital media Guidance notes on the management of adverse events and product complaints from digital media ABPI Pharmacovigilance
Questions and answers on preparation, management and assessment of periodic safety update reports (PSURs)
") 18 April 2013 EMA/CVMP/PhVWP/126661/2009-Rev.3 Committee for Medicinal Products for Veterinary Use Questions and answers on preparation, management and assessment of Output from the training of veterinary
18 April 2013 EMA/CVMP/PhVWP/126661/2009-Rev.3 Committee for Medicinal Products for Veterinary Use Questions and answers on preparation, management and assessment of Output from the training of veterinary
Clinical Research in Mauritius
 BOARD OF INVESTMENT Clinical Research in Mauritius This document provides an informative summary of the procedure to apply for a clinical research protocol in Mauritius. Contents INTRODUCTION... 2 I. APPLICATION
BOARD OF INVESTMENT Clinical Research in Mauritius This document provides an informative summary of the procedure to apply for a clinical research protocol in Mauritius. Contents INTRODUCTION... 2 I. APPLICATION
Guideline on good pharmacovigilance practices (GVP)
") 22 June 2012 EMA/873138/2011 (superseded version) Guideline on good pharmacovigilance practices (GVP) Module VI Management and reporting of adverse reactions to medicinal products Draft finalised by the
22 June 2012 EMA/873138/2011 (superseded version) Guideline on good pharmacovigilance practices (GVP) Module VI Management and reporting of adverse reactions to medicinal products Draft finalised by the
Adopted by CHMP in November 2006. Revision 1.0 adopted by CHMP on 25 June 2009
 European Medicines Agency Post-authorisation Evaluation of Medicines for Human Use London, 25 June 2009 Doc. Ref: EMEA/359381/2009 CHMP Recommendations for the Pharmacovigilance Plan as part of the Risk
European Medicines Agency Post-authorisation Evaluation of Medicines for Human Use London, 25 June 2009 Doc. Ref: EMEA/359381/2009 CHMP Recommendations for the Pharmacovigilance Plan as part of the Risk
The European regulatory system for medicines and the European Medicines Agency
 The European regulatory system for medicines and the European Medicines Agency A consistent approach to medicines regulation across the European Union An agency of the European Union This booklet is intended
The European regulatory system for medicines and the European Medicines Agency A consistent approach to medicines regulation across the European Union An agency of the European Union This booklet is intended
VOLUME 2A Procedures for marketing authorisation CHAPTER 1 MARKETING AUTHORISATION. June 2013
 EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Health systems and products Medicinal products Revision 4 NOTICE TO APPLICANTS VOLUME 2A Procedures for marketing authorisation CHAPTER 1 MARKETING
EUROPEAN COMMISSION HEALTH AND CONSUMERS DIRECTORATE-GENERAL Health systems and products Medicinal products Revision 4 NOTICE TO APPLICANTS VOLUME 2A Procedures for marketing authorisation CHAPTER 1 MARKETING
Consultation Paper. Draft Regulatory Technical Standards on the content of resolution plans and the assessment of resolvability EBA/CP/2014/16
 EBA/CP/2014/16 9 July 2014 Consultation Paper Draft Regulatory Technical Standards on the content of resolution plans and the assessment of resolvability Contents 1. Responding to this Consultation 3 2.
EBA/CP/2014/16 9 July 2014 Consultation Paper Draft Regulatory Technical Standards on the content of resolution plans and the assessment of resolvability Contents 1. Responding to this Consultation 3 2.
Opinion/ Commission. Notification. Decision. Issued 2 / affected 3 amended on. 06/07/2015 SmPC, Labelling and PL
 Procedural steps taken and scientific information after the authorisation Application Scope Opinion/ Commission Product Summary number Notification Decision Information 1 issued on Issued 2 / affected
Procedural steps taken and scientific information after the authorisation Application Scope Opinion/ Commission Product Summary number Notification Decision Information 1 issued on Issued 2 / affected
Guidance notes. for Patient Safety and Pharmacovigilance in Patient Support Programmes
 Guidance notes for Patient Safety and Pharmacovigilance in Patient Support Programmes 9 May 2011 Approval Status Authors The ABPI Pharmacovigilance Expert Network Change History N/A Approval Date 9 May
Guidance notes for Patient Safety and Pharmacovigilance in Patient Support Programmes 9 May 2011 Approval Status Authors The ABPI Pharmacovigilance Expert Network Change History N/A Approval Date 9 May
A 4.5 Validity Period of a Marketing Authorisation
 104 A 4.5 Examples of situations in which the notification procedure according to Art. 61(3) of the Community Code will apply are as follows: Minor changes to the package leaflet resulting from readability
104 A 4.5 Examples of situations in which the notification procedure according to Art. 61(3) of the Community Code will apply are as follows: Minor changes to the package leaflet resulting from readability
Questions and answers on post approval change management protocols
 30 March 2012 EMA/CHMP/CVMP/QWP/586330/2010 Committee for Medicinal Products for Human Use (CHMP) Questions and answers on post approval change management protocols Draft agreed by CHMP / CVMP Quality
30 March 2012 EMA/CHMP/CVMP/QWP/586330/2010 Committee for Medicinal Products for Human Use (CHMP) Questions and answers on post approval change management protocols Draft agreed by CHMP / CVMP Quality
Emerging Device Topics for Regulatory Consideration.. Janine Jamieson May 2015
 Emerging Device Topics for Regulatory Consideration. Janine Jamieson May 2015 Disclaimer These are my personal views and not necessarily those of MHRA as an organisation. 2 European regulation of combination
Emerging Device Topics for Regulatory Consideration. Janine Jamieson May 2015 Disclaimer These are my personal views and not necessarily those of MHRA as an organisation. 2 European regulation of combination
Questions and Answers to the Annual Safety Report
 Questions and Answers to the Annual Safety Report Frequently asked questions regarding the Development Safety Update Report (DSUR) Question 1 DSUR Start Stop 1.1 When to start preparing and where to submit
Questions and Answers to the Annual Safety Report Frequently asked questions regarding the Development Safety Update Report (DSUR) Question 1 DSUR Start Stop 1.1 When to start preparing and where to submit
End of consultation (deadline for comments) 14 October 2009. Adoption by Committee for advanced therapies 15 October 2010
 14 October 2009. Adoption by Committee for advanced therapies 15 October 2010") 15 October 2010 EMA/CAT/418458/2008/corr. Committee for advanced therapies (CAT) Procedural advice on the certification of quality and nonclinical data for small and medium sized enterprises developing
15 October 2010 EMA/CAT/418458/2008/corr. Committee for advanced therapies (CAT) Procedural advice on the certification of quality and nonclinical data for small and medium sized enterprises developing
The EU portal and database
 The EU portal and database ECPC General Assembly - 20 June 2015 Presented by Laura Pioppo Clinical and Non-clinical Compliance Service An agency of the European Union Table of contents Legal basis CT programme
The EU portal and database ECPC General Assembly - 20 June 2015 Presented by Laura Pioppo Clinical and Non-clinical Compliance Service An agency of the European Union Table of contents Legal basis CT programme
A critical review of the current. marketing authorisation transfer procedure. in Europe
 A critical review of the current marketing authorisation transfer procedure in Europe Wissenschaftliche Prüfungsarbeit zur Erlangung des Titels Master of Drug Regulatory Affairs der Mathematisch-Naturwissenschaftlichen
A critical review of the current marketing authorisation transfer procedure in Europe Wissenschaftliche Prüfungsarbeit zur Erlangung des Titels Master of Drug Regulatory Affairs der Mathematisch-Naturwissenschaftlichen
Electronic Payment Schemes Guidelines
 BANK OF TANZANIA Electronic Payment Schemes Guidelines Bank of Tanzania May 2007 Bank of Tanzania- Electronic Payment Schemes and Products Guidleness page 1 Bank of Tanzania, 10 Mirambo Street, Dar es
BANK OF TANZANIA Electronic Payment Schemes Guidelines Bank of Tanzania May 2007 Bank of Tanzania- Electronic Payment Schemes and Products Guidleness page 1 Bank of Tanzania, 10 Mirambo Street, Dar es
White Paper The EU Clinical Trials Regulation Main Changes and Challenges
 White Paper The EU Clinical Trials Regulation Main Changes and Challenges Table of Contents 1. Introduction... 3 2. Main Changes and Associated Challenges... 4 2.1 Procedure for Initial Authorisation...
White Paper The EU Clinical Trials Regulation Main Changes and Challenges Table of Contents 1. Introduction... 3 2. Main Changes and Associated Challenges... 4 2.1 Procedure for Initial Authorisation...
Patient Support Programs and Market Research Programs in Pharmacies: Managing Safety Information
 Patient Support Programs and Market Research Programs in Pharmacies: Managing Safety Information Suzete Costa Pharm D, MPH Executive Director of the Centre for Health Evaluation & Research (CEFAR) National
Patient Support Programs and Market Research Programs in Pharmacies: Managing Safety Information Suzete Costa Pharm D, MPH Executive Director of the Centre for Health Evaluation & Research (CEFAR) National
Public Assessment Report. (Budesonide) PL 17901/0254. AstraZeneca UK Ltd
 PL 17901/0254. AstraZeneca UK Ltd") Public Assessment Report Rhinocort Hay Fever 64 micrograms, Nasal Spray (Budesonide) PL 17901/0254 AstraZeneca UK Ltd 1 LAY SUMMARY Rhinocort Hay Fever 64 micrograms, Nasal Spray (Budesonide) This is a
Public Assessment Report Rhinocort Hay Fever 64 micrograms, Nasal Spray (Budesonide) PL 17901/0254 AstraZeneca UK Ltd 1 LAY SUMMARY Rhinocort Hay Fever 64 micrograms, Nasal Spray (Budesonide) This is a
COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT. Guideline on Risk Management Systems for Medicinal Products for Human Use
 DRAFT. Guideline on Risk Management Systems for Medicinal Products for Human Use") European Medicines Agency Evaluation of Medicines for Human Use London, 6 September 2005 Doc. Ref. EMEA/CHMP/96268/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT Guideline on Risk Management
European Medicines Agency Evaluation of Medicines for Human Use London, 6 September 2005 Doc. Ref. EMEA/CHMP/96268/2005 COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) DRAFT Guideline on Risk Management
Risk management plans an overview
 Risk management plans an overview Elspeth Kay Director, Risk Management Plan Evaluation Section, Post-marketing Surveillance Branch, Monitoring and Compliance Division, TGA ARCS-TGA workshop 12 March 2015
Risk management plans an overview Elspeth Kay Director, Risk Management Plan Evaluation Section, Post-marketing Surveillance Branch, Monitoring and Compliance Division, TGA ARCS-TGA workshop 12 March 2015
Health and Social Care Act 2012
 Health and Social Care Act 2012 CHAPTER 7 Explanatory Notes have been produced to assist in the understanding of this Act and are available separately 44.75 Health and Social Care Act 2012 CHAPTER 7 CONTENTS
Health and Social Care Act 2012 CHAPTER 7 Explanatory Notes have been produced to assist in the understanding of this Act and are available separately 44.75 Health and Social Care Act 2012 CHAPTER 7 CONTENTS
SANITARY AND PHYTOSANITARY MEASURES (SPS)
") TEXTUAL PROPOSAL SANITARY AND PHYTOSANITARY MEASURES (SPS) Article 1 Scope and coverage This Chapter applies to all SPS measures that may, directly or indirectly, affect trade between the Parties. This
TEXTUAL PROPOSAL SANITARY AND PHYTOSANITARY MEASURES (SPS) Article 1 Scope and coverage This Chapter applies to all SPS measures that may, directly or indirectly, affect trade between the Parties. This
THE FIRST SCHEDULE (See rule 7) Table I - FEES PAYABLE
 Table I - FEES PAYABLE") Number of entry On what payable Number of the relevant Form THE FIRST SCHEDULE (See rule 7) Table I - FEES PAYABLE Natural For e-filing Small entity, alone or with natural Others, alone or with natural
Number of entry On what payable Number of the relevant Form THE FIRST SCHEDULE (See rule 7) Table I - FEES PAYABLE Natural For e-filing Small entity, alone or with natural Others, alone or with natural
REACH. Scope REGISTRATION. The Current EU Chemicals Policy REACH
 Introduction to the New Chemicals Policy REACH REACH and Developing Countries Brussels 28-29 October 2004 Eva Sandberg DG Environment, European Commission The Current EU Chemicals Policy Problems Existing
Introduction to the New Chemicals Policy REACH REACH and Developing Countries Brussels 28-29 October 2004 Eva Sandberg DG Environment, European Commission The Current EU Chemicals Policy Problems Existing
LEMSIP MAX DAY & NIGHT COLD & FLU RELIEF CAPSULES. (paracetamol, caffeine and phenylephrine hydrochloride)
") LEMSIP MAX DAY & NIGHT COLD & FLU RELIEF CAPSULES (paracetamol, caffeine and phenylephrine hydrochloride) PL 00063/0529 UKPAR TABLE OF CONTENTS Lay Summary Page 2 Scientific Discussion Page 5 Summary of
LEMSIP MAX DAY & NIGHT COLD & FLU RELIEF CAPSULES (paracetamol, caffeine and phenylephrine hydrochloride) PL 00063/0529 UKPAR TABLE OF CONTENTS Lay Summary Page 2 Scientific Discussion Page 5 Summary of