How To Write A Binder Tab
|
|
|
- Hubert McKinney
- 5 years ago
- Views:
Transcription
1 Tool Summary Sheet Tool: Extramural Essential Documents Binder/File Tabs Purpose: To provide an organizational framework and guidance for filing paper versions of essential study documents (or referencing location of an electronically stored file) and to provide a cover page with a description of the required contents for each binder section Audience/User: Study coordinators or individuals responsible for establishing the Essential Documents Binder/File (synonyms -- Investigator Binder, Regulatory Binder, Investigational Site File [ISF] or Study Binder) Best Practice Recommendations: Details: This document clarifies the standard content of the Binder It is the responsibility of the Investigator to ensure compliance with GCP, IRB, and applicable regulatory requirements This document serves as a template and may be modified for studyspecific needs/requirements Store items in reverse chronological order, with the newest items within a section placed at the front of the section Use the requirements note at the top of each binder tab to determine if that section is required for your study Multi-site studies The lead site may choose to customize the binder tabs for the study and provide to all participating sites Electronic documents The recommendation is to store paper copies of documents in the binder. However, if you elect to use only electronic copies of particular documents, the following guidelines should be observed. o Either a) place a paper placeholder in the relevant location of the binder that directs an individual to the electronic location, OR b) place a paper placeholder in one location in the binder that includes a list of all documents that are stored only in electronic format along with the specific electronic path for each item (relevant tool: Essential Documents Storage Location Table). o Electronic-only documents should be limited to documents that a) are easily accessible by site staff; b) an inspector, auditor, or clinical monitor can be provided with easy access to the relevant electronic materials during a site visit; and c) the electronic location is controlled, regularly backed up, and is not in danger of disappearing or changing in the foreseeable future. o For correspondence, sites may want to include clarification in the binder that will be archived to a permanent storage medium on a particular schedule (specify in documentation) and the media will be stored in the binder or an easily accessible location. References: Good Clinical Practice (E6) Section 8.1, 8.2, 8.3, 8.4 v Page 1 of 24
![responsible for establishing the Essential Documents Binder/File (synonyms -- Investigator Binder, Regulatory Binder, Investigational Site File [ISF] or Study Binder) Best Practice Recommendations:](/docs-images/43/4854771/images/page_1.jpg "Details: This document clarifies the standard content of the Binder It is the responsibility of the Investigator to ensure compliance with GCP, IRB, and applicable regulatory requirements This")
2 Tool Revision History: Version Number Date Summary of Revisions Made: JUL2010 First published version MAY2011 Tool Summary Sheet title adjusted; remains v4.0 and footer version date remains the same SEP2011 Revisions incorporated and requirements notes added OCT2011 Medical license references revised and IRB Registration marked as optional JUL2012 Administrative edits and changes to reflect updates to Intramural Binder Tabs SEP2014 Removed IoR tab and revised Signed Consent Documents tab; updated hyperlinks; and completed administrative/clarifying edits v Page 2 of 24

3 Extramural Binder/File Tabs Introduction The following tabs are recommended for use in the Essential Documents Binder/File for extramural studies. This document serves as a template and may be modified for study-specific requirements. Documents should be filed in reverse chronological order within each tab. It is the responsibility of the investigator to ensure compliance with GCP and applicable regulatory requirements. To access the sample templates and tools included in the Extramural Binder/File Tabs, please visit the NIDCR Toolkit for Clinical Researchers website: v Page 3 of 24

4 Required for both observational and interventional clinical research studies Protocol and Amendments This section should include a copy of each: IRB-approved protocol Signed PI protocol signature page IRB-approved protocol amendment Signed PI protocol amendment signature page If a protocol was not submitted or approved by the IRB, a memo to file needs to be generated to explain the surrounding circumstances and the PI needs to sign and date the document. Link to NIDCR Protocol Template Tools: v Page 4 of 24

5 Required for both observational and interventional clinical research studies IRB-Approved Consent Documents This section should include a copy of: All IRB-approved /stamped consent documents A version number and date should be on each consent document. An expiration date of the consent document on the actual document is preferable, but cross-reference to the IRB approval letter of the protocol may be required. If applicable, the section should also include a copy of: All IRB-approved assent forms All IRB-approved short form consents for non-english languages* *The short form consent for non-english languages should be used for a single subject who may be illiterate, blind, or otherwise unable to read the consent document. This should be used when the full consent document has to be read or translated for subject. v Page 5 of 24

6 Required for both observational and interventional clinical research studies IRB Documentation This section should include: Federalwide Assurance (FWA) Number IRB Registration (optional) Link to OHRP Database (FWA and IRB Registration): v Page 6 of 24
: http://ohrp.cit.nih.gov/search/irbdtl.aspx v8.")
7 Required for both observational and interventional clinical research studies IRB Approvals and Correspondence This section should include a copy of: Approval letters (e.g., protocol, protocol amendment(s), consent documents, continuing review, etc.) Correspondence related to contingent approvals or stipulations Original IRB application/submission IRB correspondence Progress reports If applicable, the section should also include a copy of: Approval letters for approved assent form Approval letters for short form consent for non-english languages* Submission/acknowledgement of Investigator s Brochure Approval letter/approved advertisement or recruitment materials Approval letter/approved written educational or other materials provided to study subjects *The short form consent for non-english languages should be used for a single subject who may be illiterate, blind, or otherwise unable to read the consent document. This should be used when the full consent document has to be read or translated for subject. Link to Informed Consent Checklist: v Page 7 of 24

8 Required for both observational and interventional clinical research studies Investigator Qualification Documentation This section should include: Current Curriculum Vitae (CV) and/or other relevant dated documentation (e.g., biosketch) for all investigators A clinical (dental, medical, etc.) license for the Principal Investigator and each sub-investigator, if licensed CVs may be updated if an investigator s qualifications increase or change during the course of the study. Do not remove expired CVs as they demonstrate qualification for the entire duration of the study. Licenses should be filed behind the corresponding investigator s CV. Do not remove expired licenses. The investigators must be actively licensed in the state in which the study is conducted. The name on the license must correspond to the name on the investigator s CV and the 1572, if applicable. v Page 8 of 24

9 Required for interventional clinical studies using a drug, biologic, or device Investigator s Brochure (For any drug/product under investigation) For studies that involve administration of investigational drugs/products, this section should include: Investigator s Brochure(s) (IB), or equivalent Or Package Insert. Include labeling for approved medications. The purpose of this document is to provide information on the mechanism of action, possible risks and adverse reactions, and the expected adverse reactions associated with the previous use of the drug/product. If the package insert or the Investigator s Brochure is amended during the trial or is updated, it should be included here. v Page 9 of 24

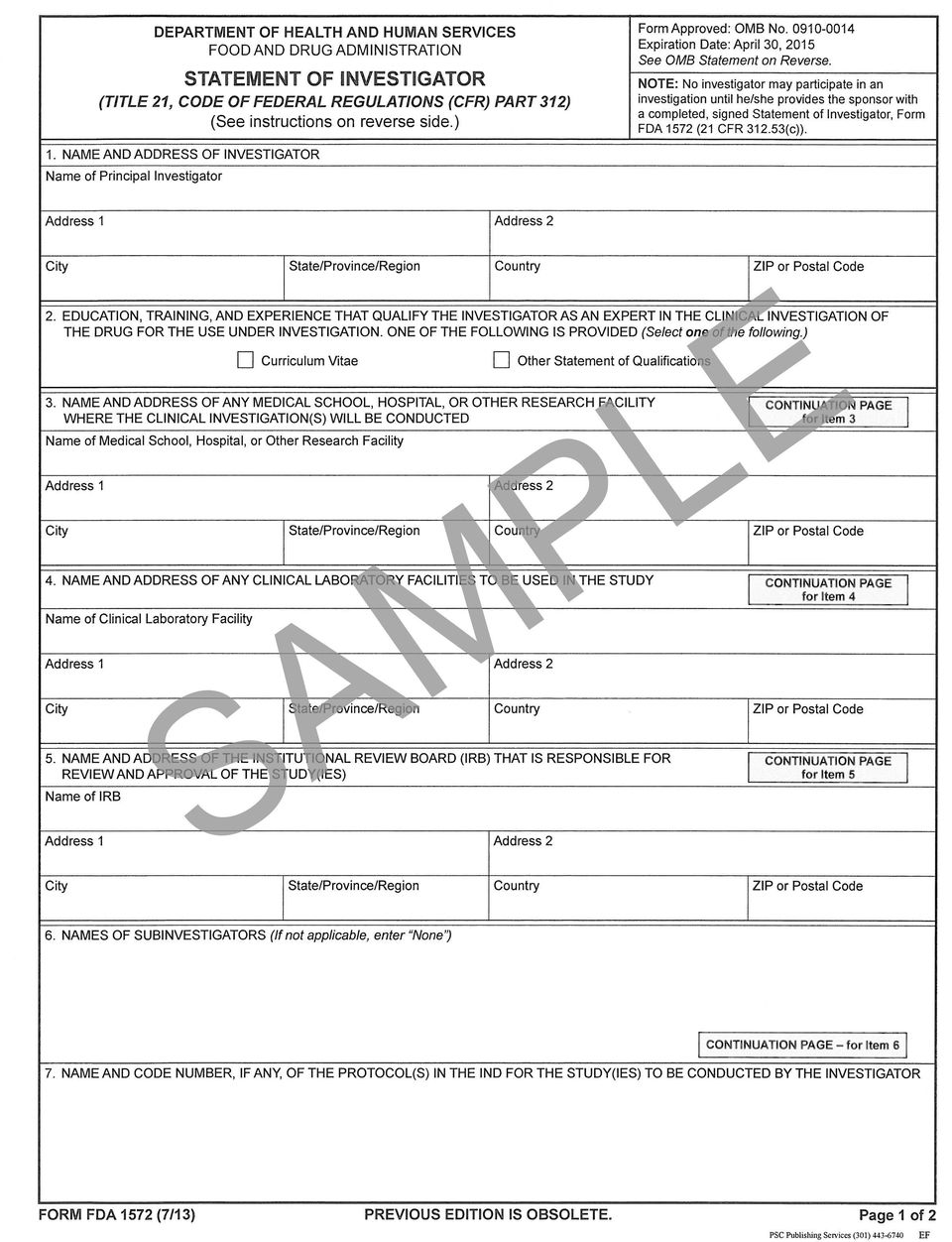
10 Required for clinical studies regulated by FDA under IND FDA Form 1572 and 1571 FDA Form 1572 for IND studies when: A study involves an investigational drug OR The study sponsor requests it FDA Form 1571 for investigator initiated INDs: Document required when an investigator is applying for an IND; part of the submission packet to the FDA Instructions for Forms 1571 and 1572: saredevelopedandapproved/approvalapplications/investigational NewDrugINDApplication/ucm htm Forms 1571 and 1572 can be downloaded from: default.htm v Page 10 of 24

11 E L S P M A
12 E L S P M A

13 E L S P M A

14 E L S P M A

15 SAMPLE

16 Required for clinical studies regulated by FDA under IND or IDE Financial Disclosure Forms This section should include: Signed Financial Disclosure Forms (FDF) for the Principal Investigator and sub-investigator(s) listed on the 1572 The names of the Principal Investigator and subinvestigator(s) should match the names listed on the The protocol title and number should match the title and number listed on the If any of the five financial interest questions are checked Yes, a statement addressing the nature and amount of the interest, arrangement, or payment must be attached to the FDF. Appropriate identifiers, i.e., protocol number and investigator name, must be included on each document included in the submission. This FDA form is required for any clinical study submitted in a marketing application that the applicant or FDA relies on to establish that the product is effective, and any study in which a single investigator makes a significant contribution to the demonstration of safety. Link for additional information: es/ucm pdf v Page 11 of 24

17 Financial Disclosure This information below is provided for the following clinical study. (Please print or type, include attachments if necessary. Maintain a copy in the official study file.) Protocol Investigator/Subinvestigator Site Protocol Title Protocol # Investigational Product(s) Pharmaceutical Co./Manufacturer(s) If answer is YES to any of the below please attach a statement addressing the nature and amount of the interest, arrangement or payment. Yes No Do you, your spouse or dependent children have a financial arrangement with the above named Pharmaceutical Company, whereby the value of compensation to you, your spouse or dependent children could be influenced by the outcome of the study? This includes compensation that could be greater for a favorable clinical result, compensation in the form of an equity interest in the above named Pharmaceutical Company, or compensation tied to sales of the product tested in the above study such as a royalty interest. Yes No Do you, your spouse or dependent children have a proprietary interest in the above named Investigational Product, such as patent rights or rights under a patent, trademark, copyright or licensing agreement? Yes No Do you, your spouse or dependent children, or any of you combined, have a significant equity in the above named Pharmaceutical Company, such as an ownership interest, stock options or any other financial interest whose value cannot be readily determined through reference to public prices, or any equity interest in the above named Pharmaceutical Company, (if it is a publicly traded organization) exceeding $50,000, or any combination of these? Yes No Have you, your spouse or dependent children, or any of you combined, received payments from the above named Pharmaceutical Company in excess of $25,000, exclusive of the costs of conducting the clinical studies, such as honoraria, a grant or grants to fund ongoing research, compensation in the form of equipment, or retainers for ongoing consultation? Yes No Have you, your spouse or dependent children had interests in a Pharmaceutical Company with a product that is in competition with the above named Investigational Product? To the best of my knowledge, the information provided above is correct and complete. I understand that I am obligated to amend this statement during the conduct of the clinical studies listed above or during one year after the studies have been completed if there is any change in this information. Signature of Investigator Disclosures should be retained in the Investigator Regulatory Binder. Date Version September 2010 Page 2 of 2

18 Required for both observational and interventional clinical research studies This section should include: Study Communication A copy of all communication relative to the conduct of the protocol and agreements with other scientific collaborators, industry, and scientific directors, such as Material Transfer Agreement and Data Sharing Agreement. (Financial documents should not be included.) Important decisions regarding study conduct, such as Memos to File FDA Correspondence (see Regulatory Document History Log) All printed communication (i.e., ) needs to be signed and dated by the individual printing and storing the document. Communication about subject treatment/clinical care, protocol deviations, and study drug dosing should immediately be printed and stored in this tab. correspondence may be saved to a compact disc (CD) for electronic storage and noted in this section Electronic media must be a permanent media, and must be appropriately secured and approved (i.e., password protected). If saved to a CD or other electronic storage media, a memo to file needs to be generated describing the types of on the electronic media, the start and stop dates of the correspondence, and the signature and date of the individual creating the CD and writing the memo to file. If a study team member receives a new computer or if a newer version of the provider is used, it is highly recommended to create the CD and the memo to file at the time of the transfer to prevent any important study communication from being lost in the transition. v Page 12 of 24

19 Regulatory Document History Log Investigator Name: Protocol: IND Number: List all documents submitted to the FDA. Date of Correspondence Type of Correspondence (i.e., submission, contact report, etc.) Serial Number (If applicable) Description Version Page Check if final page of log:
 Serial Number (If applicable) Description Version 4.")
20 Required for both observational and interventional clinical research studies Delegation of Responsibilities (DoR) Log This section should include: An ongoing log that lists all study personnel and their specific responsibilities, signatures, initials, and obligation (start/stop) dates Any changes in site study personnel require an update to the DoR. v Page 13 of 24
 dates Any changes in site study personnel require an update to the DoR. v8.")
21 Delegation of Responsibilities Log Investigator Name: Protocol: Site Number: List staff to whom the Principal Investigator (PI) has delegated significant study-related duties. Name Responsibilities* Initials Signature Start Date End Date PI Initials/Date By initialing above, I, the PI, declare that during the conduct of the above study, I have delegated the following study-related activities: *Responsibilities Legend 1. Administer Consent 2. Screen Subjects 3. Obtain Medical History 4. Perform Physical Exam 5. Determine Eligibility 6. Randomize Subjects 7. Dispense Study Drug 8. Drug Accountability 9. Assess Adverse Events 10. Complete Source Documents 11. Complete Study Forms 12. Provide Discharge Instructions 13. Make Follow-up Phone Calls 14. Query Management Signature of Principal Investigator: Date: Version Page Check if final page of log:
22 Required for both observational and interventional clinical research studies Clinical Research and Study Training This section should include the following documents for all key personnel (investigators, coordinators): Educational completion certificates for Human Subjects Protection training Documentation of study related training All key personnel working on NIH grants and contracts involving human research participants are required to complete training in Human Subject Protections. NIH has a free web-based training that satisfies this requirement. Other free, optional web-based trainings that are recommended include: Good Clinical Practices: If a certificate is not available at the end of a required training module, enter the appropriate documentation in the site Training Log. Site-specific training: Consult your IRB or institution for training requirements. Consider use of a central training binder to store non-studyspecific training documentation for all study team members. If this strategy is used, file a signed and dated memo to file explaining where these training documents are stored. v Page 14 of 24
23 Training Log Investigator Name: Protocol: Site Number: Printed Name Signature Title of Training Date of Training Version Page Check if final page of log:
24 Required for both observational and interventional clinical research studies Screening/Enrollment Log This section should include: A log without identifying information that lists subjects who were screened (including screen failures) and enrolled in the study Subjects may be tracked separately on logs, such as a coded list with a key. Note: If screening and enrollment information is entered into an electronic data capture (EDC) system, please include a memo explaining this process. v Page 15 of 24
25 Site Screening and Enrollment Log Investigator Name: Protocol: Site Number: Subject ID Date of Consent Version of Consent Date Screened Eligible for Enrollment? Ineligibility Reason (if applicable) Version Page Check if final page of log:
26 Required for both observational and interventional clinical research studies Signed Consent Documents This section should include: All original signed consent documents OR, if signed consent documents are kept in a separate binder or in the subject s medical or dental record: A signed and dated memo to file explaining where signed consent documents are stored and the reason for separate storage The signed consent document must be retained even if a subject withdraws consent or fails the screening process. v Page 16 of 24
27 Required for interventional clinical studies using a study product for research Study Product Records This section should include: Documentation of study product (drug, biologic, vaccine) disposition and accountability, or memo as to where records are located (e.g., Research Pharmacy) and who is maintaining accountability logs For masked clinical studies, it is recommended that study product accountability records be filed in the research pharmacy to maintain the masking. v Page 17 of 24
28 Investigational Product Accountability Log: Stock Record Name of Institution: Investigator Name: Protocol No.: Protocol Title: Product Name: Manufacturer: Dose Form and Strength: Dispensing Area: Line No. Date Dispensed To / Received From Dose Quantity Dispensed and/or Received Ex. 15Feb2012 Manufacturer 10 mg tabs 1. Balance Forward / Balance Lot No. Recorder s Initials JAD Version Page Check if final page of log:
29 Investigational Product Accountability Log: Subject Record Name of Institution: Investigator Name: Protocol No.: Protocol Title: Product Name: Manufacturer: Dose Form and Strength: Dispensing Area: Line No. Date Subject ID Number Subject s Initials Dose Quantity Dispensed and/or Received Ex. 15Feb ABC 10 mg tabs 1. Balance Forward / Balance Lot No. Recorder s Initials JAD Version Page Check if final page of log:
30 Required for both observational and interventional studies using clinical labs as a study procedure Local Clinical Lab Certificates/Reference Ranges For studies that utilize clinical laboratories for specimen testing, this section should include: Lab reference ranges if the reference range is not included on the lab form A copy of certifications or accreditations (CAP, CLIA, or State certificate) or a memo indicating that the laboratory maintains CLIA certification v Page 18 of 24
31 Required for both observational and interventional clinical studies collecting clinical samples Specimen Tracking Log This section should include: A log of research samples, which can include the type of specimen, purpose of storage, location of storage (e.g., freezer #, shelf #, and location, box 3), and link to subject ID number. If applicable, the log should be modified to track if consent for future use was obtained or withdrawn. v Page 19 of 24
32 Specimen Tracking Log Investigator Name: Protocol: Site Number: Visit Specimen Name/Type Specimen ID (Accession #) Date Collected Date Shipped Tracking # Receiving Lab Date Received Comments Version Page Check if final page of log:
33 Required for both observational and interventional clinical research studies Unanticipated Problems This section should include a copy of each: Unanticipated problem report v Page 20 of 24
34 Required for both observational and interventional clinical studies that are more than minimal risk Serious Adverse Events (SAEs) This section should include a copy of each: Serious Adverse Event form or memo Dear Doctor letter and IND safety report v Page 21 of 24
35 Required for both observational and interventional clinical research studies Protocol Deviations This section should include: A copy of each Protocol Deviation form or memo, or a memo to file indicating if/where they are maintained electronically (e.g., in the clinical database) Protocol Deviation Tracking Log Requirements for reporting protocol deviations are specific to each local IRB; review the requirements to make sure that they are followed appropriately. v Page 22 of 24
36 Protocol Deviation Tracking Log Protocol ID/Number: Protocol Title (Abbreviated): Site Name/Number: Principal Investigator: Page number [1]: Ref No Subject ID Date of Deviation Date Identified Deviation Identified By Deviation Description Dev. Type [2] Resulted in AE (Yes/No) Did Subject Continue in Study? SAMPLE Meets IRB Reporting Req. (Yes/No) IRB Reporting Date Action Taken (if any) Impact [3] Initials [4] Form Version Check if final page of log:
37 Form Instructions: [1] Each page should be separately numbered to allow cross-referencing (e.g., deviation #2 on p. 7) [2] Deviation Type: (A-J) See codes below Enter the appropriate deviation code from the list. Protocol Deviation Codes: A Consent Procedures B Inclusion/Exclusion Criteria C Concomitant Medication/Therapy D Laboratory Assessments/Procedures E Study Procedures F Serious Adverse Event Reporting/Unanticipated Adverse Device Effect G Randomization Procedures/Study Drug Dosing H Visit Schedule/Interval I Efficacy Ratings J Other [3] Impact: (A-D) See codes below Enter the appropriate impact code from the list. Impact Codes: A Study Validity B Safety C No Impact D Outcome Measures [4] Insert the initials of the person completing the log entry. SAMPLE Version
38 Required for both observational and interventional clinical research studies Clinical Site Monitoring Visits This section should include: A site visit log signed by the clinical site monitor(s) at each visit A copy of each visit s correspondence, which can include the confirmation letter, agenda, follow-up letter, and all attachments v Page 23 of 24
39 Monitoring Visit Log Investigator Name: Protocol: Site Number: Name Signature Purpose of Visit Date of Visit Version Page Check if final page of log:
40 As needed for both observational and interventional clinical research studies Other This section should include: Other important study documents, such as Certificates of Confidentiality, literature or publications, Technology Transfer Agreement, submissions to Radiation Safety Committee, Radiation Safety Committee approvals, submissions to Office of Biotechnology Activities, etc. v Page 24 of 24
The Regulatory Binder/Trial Master File: Essential Records for the Conduct of a Clinical Trial
 The Regulatory Binder/Trial Master File: Essential Records for the Conduct of a Clinical Trial Clinical Research Operations & Regulatory Support (CRORS) Ann Glasse, RN, BSN, MBA Director-CRORS Objectives
The Regulatory Binder/Trial Master File: Essential Records for the Conduct of a Clinical Trial Clinical Research Operations & Regulatory Support (CRORS) Ann Glasse, RN, BSN, MBA Director-CRORS Objectives
The Study Site Master File and Essential Documents
 The Study Site Master File and Essential Documents Standard Operating Procedure Office of Health and Medical Research Queensland Health SOP reference: 002 Version number: 1 Effective date: 01 June 2010
The Study Site Master File and Essential Documents Standard Operating Procedure Office of Health and Medical Research Queensland Health SOP reference: 002 Version number: 1 Effective date: 01 June 2010
Comprehensive Study Documents List (Biomedical Studies)
") Comprehensive Study Documents List (Biomedical Studies) Investigators conducting human subjects research must maintain study documents in adherence to federal and state regulations, USC policies, and good
Comprehensive Study Documents List (Biomedical Studies) Investigators conducting human subjects research must maintain study documents in adherence to federal and state regulations, USC policies, and good
Essential Documents for the Conduct of a Clinical Trial. Debra Dykhuis Associate Director RSO
 Essential Documents for the Conduct of a Clinical Trial Debra Dykhuis Associate Director RSO Introduction Rationale for choosing this topic AHC movement toward setting GCP (Good Clinical Practice) guidelines
Essential Documents for the Conduct of a Clinical Trial Debra Dykhuis Associate Director RSO Introduction Rationale for choosing this topic AHC movement toward setting GCP (Good Clinical Practice) guidelines
Study Start-Up SS-204.01. STANDARD OPERATING PROCEDURE FOR Site Initiation Visit (SIV)
") Study Start-Up SS-204.01 STANDARD OPERATING PROCEDURE FOR Site Initiation Visit (SIV) Approval: Nancy Paris, MS, FACHE President and CEO 08 March 2012 (Signature and Date) Approval: Frederick M. Schnell,
Study Start-Up SS-204.01 STANDARD OPERATING PROCEDURE FOR Site Initiation Visit (SIV) Approval: Nancy Paris, MS, FACHE President and CEO 08 March 2012 (Signature and Date) Approval: Frederick M. Schnell,
RECOMMENDATION ON THE CONTENT OF THE TRIAL MASTER FILE AND ARCHIVING
 RECOMMENDATION ON THE CONTENT OF THE TRIAL MASTER FILE AND ARCHIVING July 2006 TABLE OF CONTENTS Page 1. Introduction 2 2. Scope 2 3. Documents to be archived 2 4. Quality of essential documents 10 5.
RECOMMENDATION ON THE CONTENT OF THE TRIAL MASTER FILE AND ARCHIVING July 2006 TABLE OF CONTENTS Page 1. Introduction 2 2. Scope 2 3. Documents to be archived 2 4. Quality of essential documents 10 5.
Subject: No. Page PROTOCOL AND CASE REPORT FORM DEVELOPMENT AND REVIEW Standard Operating Procedure
 703 1 of 11 POLICY The Beaumont Research Coordinating Center (BRCC) will provide advice to clinical trial investigators on protocol development, content and format. Upon request, the BRCC will review a
703 1 of 11 POLICY The Beaumont Research Coordinating Center (BRCC) will provide advice to clinical trial investigators on protocol development, content and format. Upon request, the BRCC will review a
Regulatory Binder Instructions 25 April 2016
 Regulatory Binder Instructions 25 April 2016 Instructions This Regulatory Binder is available to help study sites achieve and maintain regulatory compliance and adhere to high standards of practice in
Regulatory Binder Instructions 25 April 2016 Instructions This Regulatory Binder is available to help study sites achieve and maintain regulatory compliance and adhere to high standards of practice in
Clinical Investigator Inspections and FDA-483 Observations
 Clinical Investigator Inspections and FDA-483 Observations Nancy A. Bellamy, Investigator Bioresearch Specialist/ BIMO Coordinator FDA Detroit District Office October 2, 2013 Objectives Background on FDA
Clinical Investigator Inspections and FDA-483 Observations Nancy A. Bellamy, Investigator Bioresearch Specialist/ BIMO Coordinator FDA Detroit District Office October 2, 2013 Objectives Background on FDA
ROLES, RESPONSIBILITIES AND DELEGATION OF DUTIES IN CLINICAL TRIALS OF MEDICINAL PRODUCTS
 ROLES, RESPONSIBILITIES AND DELEGATION OF DUTIES IN CLINICAL TRIALS OF MEDICINAL PRODUCTS STANDARD OPERATING PROCEDURE NO SOP 09 DATE RATIFIED 4/7/13 NEXT REVIEW DATE 4/7/14 POLICY STATEMENT/KEY OBJECTIVES:
ROLES, RESPONSIBILITIES AND DELEGATION OF DUTIES IN CLINICAL TRIALS OF MEDICINAL PRODUCTS STANDARD OPERATING PROCEDURE NO SOP 09 DATE RATIFIED 4/7/13 NEXT REVIEW DATE 4/7/14 POLICY STATEMENT/KEY OBJECTIVES:
No. 706. Page 1 of 5. Issue Date 4/21/2014
 Subject: ROUTINE MONITORING VISITS Standard Operating Procedure Prepared By: Beaumont Research Coordinating Center, Research Institute PURPOSE Prior Issue Date 8/15/2011 No. 706 Issue Date 4/21/2014 Page
Subject: ROUTINE MONITORING VISITS Standard Operating Procedure Prepared By: Beaumont Research Coordinating Center, Research Institute PURPOSE Prior Issue Date 8/15/2011 No. 706 Issue Date 4/21/2014 Page
STANDARD OPERATING PROCEDURE FOR RESEARCH. Management of Essential Documents and Trial Folders
 STANDARD OPERATING PROCEDURE FOR RESEARCH Management of Essential Documents and Trial Folders Author Linda Ward Author s Job Title QA Coordinator Division Department Version number 2 Ref SOP/CLN/001/2
STANDARD OPERATING PROCEDURE FOR RESEARCH Management of Essential Documents and Trial Folders Author Linda Ward Author s Job Title QA Coordinator Division Department Version number 2 Ref SOP/CLN/001/2
Site Activation: Keep Your Eyes on the Prize
 Site Activation: Keep Your Eyes on the Prize Theresa Gamble, Ph.D. Senior Clinical Research Manager HPTN CORE, FHI October 2004 Slide 1 The Prize Site-Specific Protocol Activation Notice See Example Document
Site Activation: Keep Your Eyes on the Prize Theresa Gamble, Ph.D. Senior Clinical Research Manager HPTN CORE, FHI October 2004 Slide 1 The Prize Site-Specific Protocol Activation Notice See Example Document
TRIAL MASTER FILE- SPONSORED
 gsop-06-04 - Management of TMF for ENHT/ WHHT Sponsored CTIMPs Page 1 of 16 Hertfordshire Hospitals R&D Consortium Incorporating West Herts Hospitals NHS Trust and East & North Herts NHS Trust TRIAL MASTER
gsop-06-04 - Management of TMF for ENHT/ WHHT Sponsored CTIMPs Page 1 of 16 Hertfordshire Hospitals R&D Consortium Incorporating West Herts Hospitals NHS Trust and East & North Herts NHS Trust TRIAL MASTER
CNE Disclosures. To change this title, go to Notes Master
 CNE Disclosures Successful Completion: Participants must complete an evaluation form to receive a certificate of completion Contact Hours: 1 contact hour is available to those who meet the successful completion
CNE Disclosures Successful Completion: Participants must complete an evaluation form to receive a certificate of completion Contact Hours: 1 contact hour is available to those who meet the successful completion
Documentation of the Informed Consent Process. USC Office for the Protection of Research Subjects (OPRS)
") Documentation of the Informed Consent Process USC Office for the Protection of Research Subjects (OPRS) Session Overview Highlights: Purpose of Informed Consent (IC) IC Process and Documentation Witness
Documentation of the Informed Consent Process USC Office for the Protection of Research Subjects (OPRS) Session Overview Highlights: Purpose of Informed Consent (IC) IC Process and Documentation Witness
The Monitoring Visit. Denise Owensby, CCRP Sr. Clinical Research Coordinator Clinical & Translational Science Center University of California, Davis
 The Monitoring Visit Denise Owensby, CCRP Sr. Clinical Research Coordinator Clinical & Translational Science Center University of California, Davis Disclosure The information herein is not intended to
The Monitoring Visit Denise Owensby, CCRP Sr. Clinical Research Coordinator Clinical & Translational Science Center University of California, Davis Disclosure The information herein is not intended to
The Importance of Following the PROTOCOL in Clinical Trials
 The Importance of Following the PROTOCOL in Clinical Trials Presentation Objectives: Upon completion of this presentation, participants will be able to: Describe the following terms: Protocol, Protocol
The Importance of Following the PROTOCOL in Clinical Trials Presentation Objectives: Upon completion of this presentation, participants will be able to: Describe the following terms: Protocol, Protocol
12.0 Investigator Responsibilities
 v. 5.13.13 12.0 Investigator Responsibilities 12.1 Policy Investigators are ultimately responsible for the conduct of research. Research must be conducted according to the signed Investigator statement,
v. 5.13.13 12.0 Investigator Responsibilities 12.1 Policy Investigators are ultimately responsible for the conduct of research. Research must be conducted according to the signed Investigator statement,
CLINICAL RESEARCH PROTOCOL CHECKLIST
 CLINICAL RESEARCH PROTOCOL CHECKLIST [taken from ICH GCP : Guidance for Industry, Good Clinical Practice: Consolidated Guidance, Revision 1 (R1) June 1996] ICH GCP, Section 6. CLINICAL TRIAL PROTOCOL AND
CLINICAL RESEARCH PROTOCOL CHECKLIST [taken from ICH GCP : Guidance for Industry, Good Clinical Practice: Consolidated Guidance, Revision 1 (R1) June 1996] ICH GCP, Section 6. CLINICAL TRIAL PROTOCOL AND
Quality Monitoring Checklist
 Quality Monitoring Checklist Instructions: For each task below, the Quality Monitor indicates in the appropriate column if the Monitor accomplished the task by using the following codes Yes No N/A = Monitor
Quality Monitoring Checklist Instructions: For each task below, the Quality Monitor indicates in the appropriate column if the Monitor accomplished the task by using the following codes Yes No N/A = Monitor
Medical College of Georgia SOP NUMBER: 03 INVESTIGATIONAL DRUG HANDLING Version Number: 1.0, 1.1 Effective Date: 09/12/06, 08/02/10, 3/2/11
 Effective Date: 09/12/06, 08/02/10, 3/2/11 Title: 1.0 OBJECTIVE: 1.1 This SOP describes the methods and policies for: Handling investigational drug Dispensing investigational drug 1.2. This procedure applies
Effective Date: 09/12/06, 08/02/10, 3/2/11 Title: 1.0 OBJECTIVE: 1.1 This SOP describes the methods and policies for: Handling investigational drug Dispensing investigational drug 1.2. This procedure applies
GOOD CLINICAL PRACTICE: CONSOLIDATED GUIDELINE
 ICH E6 GCP: Consolidated Guideline: Investigator 1/7 Institutional Review Board Services ICH HARMONIZED TRIPARTITE GUIDELINE E6: GOOD CLINICAL PRACTICE: CONSOLIDATED GUIDELINE 4. INVESTIGATOR 4.1 Investigator's
ICH E6 GCP: Consolidated Guideline: Investigator 1/7 Institutional Review Board Services ICH HARMONIZED TRIPARTITE GUIDELINE E6: GOOD CLINICAL PRACTICE: CONSOLIDATED GUIDELINE 4. INVESTIGATOR 4.1 Investigator's
Guidance for Industry Investigator Responsibilities Protecting the Rights, Safety, and Welfare of Study Subjects
 Guidance for Industry Investigator Responsibilities Protecting the Rights, Safety, and Welfare of Study Subjects U.S. Department of Health and Human Services Food and Drug Administration Center for Drug
Guidance for Industry Investigator Responsibilities Protecting the Rights, Safety, and Welfare of Study Subjects U.S. Department of Health and Human Services Food and Drug Administration Center for Drug
ROLE OF THE RESEARCH COORDINATOR Study Start-up Best Practices
 Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Study Start-up
Clinical and Translational Science Institute / CTSI at the University of California, San Francisco Welcome to Online Training for Clinical Research Coordinators ROLE OF THE RESEARCH COORDINATOR Study Start-up
Principal Investigator Responsibilities for Education and Social/Behavioral Researchers
 Principal Investigator Responsibilities for Education and Social/Behavioral Researchers Introduction The purpose of this module is to provide a basic understanding of the responsibilities of the principal
Principal Investigator Responsibilities for Education and Social/Behavioral Researchers Introduction The purpose of this module is to provide a basic understanding of the responsibilities of the principal
DHHS/NIH/OD/OIR/OHSRP 1/2/2015
 DHHS/NIH/OD/OIR/OHSRP 1/2/2015 The audience for this course is Principal Investigators (PIs), investigators and Research Coordinators (RCs) serving on the study team of human clinical studies and trials.
DHHS/NIH/OD/OIR/OHSRP 1/2/2015 The audience for this course is Principal Investigators (PIs), investigators and Research Coordinators (RCs) serving on the study team of human clinical studies and trials.
Guide for Research Sites Seeking Accreditation
 Guide for Research Sites Seeking Accreditation (For research sites that only conduct research and do not have their own IRBs) November 16, 2010 Purpose of the Guide The accreditation process for most research
Guide for Research Sites Seeking Accreditation (For research sites that only conduct research and do not have their own IRBs) November 16, 2010 Purpose of the Guide The accreditation process for most research
Pre-Questions. Mastering Clinical Research July 29, 2015
 Pre-Questions Mastering Clinical Research July 29, 2015 1. To be compliant with SOP 2.1 Obtaining Informed Consent for greater than minimal risk interventional clinical trial, which Licensed Professional
Pre-Questions Mastering Clinical Research July 29, 2015 1. To be compliant with SOP 2.1 Obtaining Informed Consent for greater than minimal risk interventional clinical trial, which Licensed Professional
STANDARD OPERATING PROCEDURES FOR GOOD CLINICAL PRACTICE
 STANDARD OPERATING PROCEDURES FOR GOOD CLINICAL PRACTICE UNC OB/GYN (V.1) 9.1.2014 Page i TABLE OF CONTENTS INTRODUCTION. iii ABBREVIATIONS iv GLOSSARY v LISTING OF ATTACHMENTS.... xi I. 1.0 GENERAL ADMINISTRATION
STANDARD OPERATING PROCEDURES FOR GOOD CLINICAL PRACTICE UNC OB/GYN (V.1) 9.1.2014 Page i TABLE OF CONTENTS INTRODUCTION. iii ABBREVIATIONS iv GLOSSARY v LISTING OF ATTACHMENTS.... xi I. 1.0 GENERAL ADMINISTRATION
CLINICAL DATA MONITORING PLAN (CDMoP) PROTOCOL # [0000] [TITLE]
![CLINICAL DATA MONITORING PLAN (CDMoP) PROTOCOL # [0000] [TITLE]](/thumbs/39/18344673.jpg "CLINICAL DATA MONITORING PLAN (CDMoP) PROTOCOL # [0000] [TITLE]") CLINICAL DATA MONITORING PLAN (CDMoP) PROTOCOL # [0000] [TITLE] CONTRACT RESEARCH ORGANIZATION SPONSOR [NAME] [ADDRESS] 1 TABLE OF CONTENTS 1. Purpose 3 2. References 3 3. Study Roles and Responsibilities
CLINICAL DATA MONITORING PLAN (CDMoP) PROTOCOL # [0000] [TITLE] CONTRACT RESEARCH ORGANIZATION SPONSOR [NAME] [ADDRESS] 1 TABLE OF CONTENTS 1. Purpose 3 2. References 3 3. Study Roles and Responsibilities
Information Sheet Guidance for Sponsors, Clinical Investigators, and IRBs
 Information Sheet Guidance for Sponsors, Clinical Investigators, and IRBs Frequently Asked Questions Statement of Investigator (Form FDA 1572) U.S. Department of Health and Human Services Food and Drug
Information Sheet Guidance for Sponsors, Clinical Investigators, and IRBs Frequently Asked Questions Statement of Investigator (Form FDA 1572) U.S. Department of Health and Human Services Food and Drug
Hollie Goddard Sr. IRB Coordinator McKesson Specialty Health
 Hollie Goddard Sr. IRB Coordinator McKesson Specialty Health We are responsible for acquiring, analyzing, and protecting medical information vital to providing quality patient care HIM professionals ensure
Hollie Goddard Sr. IRB Coordinator McKesson Specialty Health We are responsible for acquiring, analyzing, and protecting medical information vital to providing quality patient care HIM professionals ensure
ICH CRA Certification Guide March 2009
 ICH CRA Certification Guide March 2009 ICH CRA CERTIFICATION GUIDE... 1 GENERAL INFORMATION... 2 BENEFITS OF CERTIFICATION... 2 INDUSTRY RECOGNITION... 2 ABOUT THE EXAM... 2 CRA DEFINITION... 2 REQUIREMENTS
ICH CRA Certification Guide March 2009 ICH CRA CERTIFICATION GUIDE... 1 GENERAL INFORMATION... 2 BENEFITS OF CERTIFICATION... 2 INDUSTRY RECOGNITION... 2 ABOUT THE EXAM... 2 CRA DEFINITION... 2 REQUIREMENTS
Orientation Manual for Clinical Research Coordinators
 Orientation Manual for Clinical Research Coordinators Maine Medical Center Research Institute Page 1 of 19 Version 1 (2009) MAINE MEDICAL CENTER RESEARCH INSTITUTE Statement of Mission, Vision, Goals and
Orientation Manual for Clinical Research Coordinators Maine Medical Center Research Institute Page 1 of 19 Version 1 (2009) MAINE MEDICAL CENTER RESEARCH INSTITUTE Statement of Mission, Vision, Goals and
University of Hawai i Human Studies Program. Guidelines for Developing a Clinical Research Protocol
 University of Hawai i Human Studies Program Guidelines for Developing a Clinical Research Protocol Following are guidelines for writing a clinical research protocol for submission to the University of
University of Hawai i Human Studies Program Guidelines for Developing a Clinical Research Protocol Following are guidelines for writing a clinical research protocol for submission to the University of
Investigational Drugs: Investigational Drugs and Biologics
 : I. PURPOSE The purpose of this policy is to establish procedures for the proper control, storage, use and handling of investigational drugs and biologics to ensure that adequate safeguards are in place
: I. PURPOSE The purpose of this policy is to establish procedures for the proper control, storage, use and handling of investigational drugs and biologics to ensure that adequate safeguards are in place
CONTROLLED DOCUMENT- DO NOT COPY STANDARD OPERATING PROCEDURE. STH Investigator
 Research Department STANDARD OPERATING PROCEDURE STH Investigator Archiving of Essential Documentation Generated During Clinical Research SOP Number A127 Version Number V1.3 Effective Date Author Zoe Whiteley
Research Department STANDARD OPERATING PROCEDURE STH Investigator Archiving of Essential Documentation Generated During Clinical Research SOP Number A127 Version Number V1.3 Effective Date Author Zoe Whiteley
Clinical Investigator Training Course
 Clinical Investigator Training Course Investigator Responsibilities in Biomedical Research Covered by FDA Regulations Lisa Zimmerman Objectives Discuss Clinical Investigator Obligations according to FDA
Clinical Investigator Training Course Investigator Responsibilities in Biomedical Research Covered by FDA Regulations Lisa Zimmerman Objectives Discuss Clinical Investigator Obligations according to FDA
A Principal Investigator s Guide to Responsibilities, Qualifications, Records and Documentation of Human Research University of Kentucky
 A Principal Investigator s Guide to Responsibilities, Qualifications, Records and Documentation of Human Research University of Kentucky I. Compliance with IRB and Applicable Federal Requirements A. Investigators
A Principal Investigator s Guide to Responsibilities, Qualifications, Records and Documentation of Human Research University of Kentucky I. Compliance with IRB and Applicable Federal Requirements A. Investigators
INTERIM SITE MONITORING PROCEDURE
 INTERIM SITE MONITORING PROCEDURE 1. PURPOSE The purpose of this SOP is to describe the interim monitoring procedures conducted at Institution, according to GCP and other applicable local regulations.
INTERIM SITE MONITORING PROCEDURE 1. PURPOSE The purpose of this SOP is to describe the interim monitoring procedures conducted at Institution, according to GCP and other applicable local regulations.
Human Research Protection Program Good Clinical Practice Guidance for Investigators Investigator & Research Staff Responsibilities
 This Guidance Document is to ensure that investigators and research personnel recognize their responsibilities associated with the conduct of human subject research by outlining their responsibilities,
This Guidance Document is to ensure that investigators and research personnel recognize their responsibilities associated with the conduct of human subject research by outlining their responsibilities,
Principal Investigator and Sub Investigator Responsibilities
 Principal Investigator and Sub Investigator Responsibilities I. Purpose To define the roles and responsibilities of Principal Investigators conducting research at GRU. II. Definition The term Principal
Principal Investigator and Sub Investigator Responsibilities I. Purpose To define the roles and responsibilities of Principal Investigators conducting research at GRU. II. Definition The term Principal
Research Study Close-down and Archiving Procedures
 Title: Outcome Statement: Research Study Close-down and Archiving Procedures To inform researchers of the process for closing down research studies, retaining and storing research materials in the Trust.
Title: Outcome Statement: Research Study Close-down and Archiving Procedures To inform researchers of the process for closing down research studies, retaining and storing research materials in the Trust.
Objectives. The Paper Tells the Story
 Notes to File: An Auditor s Perspective Lorrie D. Divers, CCRP, RQAP-GCP Executive Director, Global Quality Assurance & Compliance ACM Medical Laboratory / ACM Global Central Laboratory Clinical Translation
Notes to File: An Auditor s Perspective Lorrie D. Divers, CCRP, RQAP-GCP Executive Director, Global Quality Assurance & Compliance ACM Medical Laboratory / ACM Global Central Laboratory Clinical Translation
Essential Documents for Clinical Trial Research. Centre for Health Evaluation and Outcome Sciences Manager, Clinical Research Development Office
 Essential Documents for Clinical Trial Research Erin Cherban, MSc., CCRP Centre for Health Evaluation and Outcome Sciences Manager, Clinical Research Development Office Document Examples See the following
Essential Documents for Clinical Trial Research Erin Cherban, MSc., CCRP Centre for Health Evaluation and Outcome Sciences Manager, Clinical Research Development Office Document Examples See the following
CLINICAL RESEARCH ROLES CLINICAL RESEARCH ROLESCLINICAL RESEARCH ROLES
 CLINICAL RESEARCH ROLESCLINICAL RESEARCH ROLES 1. Clinical Study Team Principal Investigator Co Principal Investigator (Co PI) Research Coordinator Study Monitor Data manage Sponsor Post Doctoral Scholar
CLINICAL RESEARCH ROLESCLINICAL RESEARCH ROLES 1. Clinical Study Team Principal Investigator Co Principal Investigator (Co PI) Research Coordinator Study Monitor Data manage Sponsor Post Doctoral Scholar
Monitoring & Auditing of Clinical Trials. Sponsored by Center for Cancer Research National Cancer Institute
 Monitoring & Auditing of Clinical Trials Sponsored by Center for Cancer Research National Cancer Institute Objectives Guidelines suggest that following the good clinical research practice of monitoring/auditing
Monitoring & Auditing of Clinical Trials Sponsored by Center for Cancer Research National Cancer Institute Objectives Guidelines suggest that following the good clinical research practice of monitoring/auditing
The role, duties and responsibilities of clinical trials personnel Monitoring: rules and recommendations
 The role, duties and responsibilities of clinical trials personnel Monitoring: rules and recommendations Maria Luisa Paoloni OPBG Clinical & Research Services Monitoring and Responsible of monitoring:
The role, duties and responsibilities of clinical trials personnel Monitoring: rules and recommendations Maria Luisa Paoloni OPBG Clinical & Research Services Monitoring and Responsible of monitoring:
Roles & Responsibilities of the Sponsor
 Roles & Responsibilities of the Sponsor Developed by Center for Cancer Research, National Cancer Institute, NIH Endorsed by the CTN SIG Leadership Group Objectives Funding for clinical research comes from
Roles & Responsibilities of the Sponsor Developed by Center for Cancer Research, National Cancer Institute, NIH Endorsed by the CTN SIG Leadership Group Objectives Funding for clinical research comes from
MINNEAPOLIS MEDICAL RESEARCH FOUNDATION CLINICAL RESEARCH STANDARD OPERATING PROCEDURES TABLE OF CONTENTS
 MINNEAPOLIS MEDICAL RESEARCH FOUNDATION CLINICAL RESEARCH STANDARD OPERATING PROCEDURES TABLE OF CONTENTS CATEGORY SOP # Clinical Research: Administrative Standard Operating Procedures (SOPs) 1.1 Outreach
MINNEAPOLIS MEDICAL RESEARCH FOUNDATION CLINICAL RESEARCH STANDARD OPERATING PROCEDURES TABLE OF CONTENTS CATEGORY SOP # Clinical Research: Administrative Standard Operating Procedures (SOPs) 1.1 Outreach
Objectives. Monitoring & Auditing of Clinical Trials. Overview. Pre-study Qualification Visit. Industry-sponsored Trials
 Objectives Monitoring & Auditing of Clinical Trials Sponsored by Center for Cancer Research National Cancer Institute Guidelines suggest that following the good clinical research practice of monitoring/auditing
Objectives Monitoring & Auditing of Clinical Trials Sponsored by Center for Cancer Research National Cancer Institute Guidelines suggest that following the good clinical research practice of monitoring/auditing
PREP Course #27: Medical Device Clinical Trial Management
 PREP Course #27: Medical Device Clinical Trial Management Presented by: Evelyn Huang Jeffrey Revello Office of Research Compliance North Shore-LIJ Health System CME Disclosure Statement The North Shore
PREP Course #27: Medical Device Clinical Trial Management Presented by: Evelyn Huang Jeffrey Revello Office of Research Compliance North Shore-LIJ Health System CME Disclosure Statement The North Shore
Guidance on IRB Continuing Review of Research
 NOTE: THIS GUIDANCE SUPERSEDES OHRP S JANUARY 15, 2007 GUIDANCE ENTITILED GUIDANCE ON CONTINUING REVIEW. CLICK HERE FOR THE JANUARY 15, 2007 GUIDANCE. Office for Human Research Protections Department of
NOTE: THIS GUIDANCE SUPERSEDES OHRP S JANUARY 15, 2007 GUIDANCE ENTITILED GUIDANCE ON CONTINUING REVIEW. CLICK HERE FOR THE JANUARY 15, 2007 GUIDANCE. Office for Human Research Protections Department of
Research Coordinator - PI s who have research coordinators or secretarial support can designate individuals to manage their IRB protocols in Mentor.
 Mentor Online IRB System IRB s require lots of documentation and managing this process can get to be a burden for both investigators and the IRB committee and administrator. The Mentor IRB system is designed
Mentor Online IRB System IRB s require lots of documentation and managing this process can get to be a burden for both investigators and the IRB committee and administrator. The Mentor IRB system is designed
Guidance to Research Ethics Committees on Initial Facility Assessment
 Guidance to Research Ethics Committees on Initial Facility Assessment Introduction One of the roles of a Research Ethics Committee is to provide an opinion on a proposed study, based on the suitability
Guidance to Research Ethics Committees on Initial Facility Assessment Introduction One of the roles of a Research Ethics Committee is to provide an opinion on a proposed study, based on the suitability
Clinical Research Professional Certification & Preparing for the CCRP Exam
 Clinical Research Professional Certification & Preparing for the CCRP Exam Signe Denmark, MS, CCRP Toni Mauney, CCRP SoCRA: Society of Clinical Research Associates SoCRA established the Certification Program
Clinical Research Professional Certification & Preparing for the CCRP Exam Signe Denmark, MS, CCRP Toni Mauney, CCRP SoCRA: Society of Clinical Research Associates SoCRA established the Certification Program
2 Applicability: Effective Date: 1/15/2010 Revised: 8/13/2010, 9/10/10, 5/9/14
 2 Applicability: Effective Date: 1/15/2010 Revised: 8/13/2010, 9/10/10, 5/9/14 NDSU research may involve the collaboration or assistance of other research institutions, schools, hospitals, clinics, private
2 Applicability: Effective Date: 1/15/2010 Revised: 8/13/2010, 9/10/10, 5/9/14 NDSU research may involve the collaboration or assistance of other research institutions, schools, hospitals, clinics, private
Web Meeting 12./13.12.2013: Study Close-out Procedures
 SiLVER Study A prospective, randomized, open-labeled trial comparing sirolimuscontaining versus mtor-inhibitor-free immunosuppression in patients undergoing liver transplantation for hepatocellular carcinoma
SiLVER Study A prospective, randomized, open-labeled trial comparing sirolimuscontaining versus mtor-inhibitor-free immunosuppression in patients undergoing liver transplantation for hepatocellular carcinoma
Version history Version number Version date Effective date 01 dd-mon-yyyy dd-mon-yyyy 02 dd-mon-yyyy dd-mon-yyyy 03 (current) dd-mon-yyyy dd-mon-yyyy
 dd-mon-yyyy dd-mon-yyyy") Trial name: HOVON xxx yyy Sponsor: HOVON Version history Version number Version date Effective date 01 dd-mon-yyyy dd-mon-yyyy 02 dd-mon-yyyy dd-mon-yyyy 03 (current) dd-mon-yyyy dd-mon-yyyy QRMP authors
Trial name: HOVON xxx yyy Sponsor: HOVON Version history Version number Version date Effective date 01 dd-mon-yyyy dd-mon-yyyy 02 dd-mon-yyyy dd-mon-yyyy 03 (current) dd-mon-yyyy dd-mon-yyyy QRMP authors
[NAME OF NATIONAL REGULATORY AUTHORITY] PROCEDURE FOR SUBMISSION CLINICAL TRIAL APPLICATIONS VACCINES AND BIOLOGICALS [COUNTRY]
![[NAME OF NATIONAL REGULATORY AUTHORITY] PROCEDURE FOR SUBMISSION CLINICAL TRIAL APPLICATIONS VACCINES AND BIOLOGICALS [COUNTRY]](/thumbs/27/12166964.jpg "[NAME OF NATIONAL REGULATORY AUTHORITY] PROCEDURE FOR SUBMISSION CLINICAL TRIAL APPLICATIONS VACCINES AND BIOLOGICALS [COUNTRY]") [TEMPLATE] [NAME OF NATIONAL REGULATORY AUTHORITY] PROCEDURE FOR SUBMISSION OF CLINICAL TRIAL APPLICATIONS OF VACCINES AND BIOLOGICALS IN [COUNTRY] 1of 19 General Procedures for clinical trial applications
[TEMPLATE] [NAME OF NATIONAL REGULATORY AUTHORITY] PROCEDURE FOR SUBMISSION OF CLINICAL TRIAL APPLICATIONS OF VACCINES AND BIOLOGICALS IN [COUNTRY] 1of 19 General Procedures for clinical trial applications
Essential Standard Operating Procedures Sample Templates Table of Contents
 Essential Standard Operating Procedures Sample Templates Table of Contents Introduction Study Conduct and Good Clinical Practice 1: JHM Training/Certification Documentation 2: Delegation of Responsibility
Essential Standard Operating Procedures Sample Templates Table of Contents Introduction Study Conduct and Good Clinical Practice 1: JHM Training/Certification Documentation 2: Delegation of Responsibility
Nova Southeastern University Institutional Review Board Policies and Procedures
 Nova Southeastern University Institutional Review Board Policies and Procedures Monitoring of Approved Research, Approval Duration, and Continuing Review Effective 03/08/2007; Revised 10/14/2010; 8/29/2011;
Nova Southeastern University Institutional Review Board Policies and Procedures Monitoring of Approved Research, Approval Duration, and Continuing Review Effective 03/08/2007; Revised 10/14/2010; 8/29/2011;
Investigator responsibilities for research conducted under the authority of the UTHSCSA Institutional Review Board (IRB)
") March 1, 2006 M E M O R A N D U M F O R R E C O R D TO: FROM: SUBJECT: Deans Department Chairs Principal Investigators Brian Herman, Ph.D. Vice President for Research Investigator responsibilities for
March 1, 2006 M E M O R A N D U M F O R R E C O R D TO: FROM: SUBJECT: Deans Department Chairs Principal Investigators Brian Herman, Ph.D. Vice President for Research Investigator responsibilities for
This policy applies to all clinical research conducted at Beaumont Health System.
 CLINICAL RESEARCH QUALITY AND PROCESS IMPROVEMENT PROGRAM 113 1 of 6 PURPOSE Prior The purpose of this policy is to provide an overview of the Clinical Research Quality and Process Improvement Program
CLINICAL RESEARCH QUALITY AND PROCESS IMPROVEMENT PROGRAM 113 1 of 6 PURPOSE Prior The purpose of this policy is to provide an overview of the Clinical Research Quality and Process Improvement Program
REGULATIONS FOR THE POSTGRADUATE DIPLOMA IN CLINICAL RESEARCH METHODOLOGY (PDipClinResMethodology)
") 452 REGULATIONS FOR THE POSTGRADUATE DIPLOMA IN CLINICAL RESEARCH METHODOLOGY (PDipClinResMethodology) (See also General Regulations) M.57 Admission requirements To be eligible for admission to the courses
452 REGULATIONS FOR THE POSTGRADUATE DIPLOMA IN CLINICAL RESEARCH METHODOLOGY (PDipClinResMethodology) (See also General Regulations) M.57 Admission requirements To be eligible for admission to the courses
Overview of Study Start Up Activities for a Clinical Trial at an Investigative Site
 University of North Texas Health Science Center UNTHSC Scholarly Repository Theses and Dissertations 11-2012 Overview of Study Start Up Activities for a Clinical Trial at an Investigative Site Prianka
University of North Texas Health Science Center UNTHSC Scholarly Repository Theses and Dissertations 11-2012 Overview of Study Start Up Activities for a Clinical Trial at an Investigative Site Prianka
STANDARD OPERATING PROCEDURE NO. CM.13-00 - 00
 STANDARD OPERATING PROCEDURE NO. CM.13-00 - 00 Version date: Effective Date: Replaces SOP No.: 1 5 January 20 13 15 February 20 13 Approved by: Date No: CM.13 00 00 Effective Date: 15 February 2013 Version
STANDARD OPERATING PROCEDURE NO. CM.13-00 - 00 Version date: Effective Date: Replaces SOP No.: 1 5 January 20 13 15 February 20 13 Approved by: Date No: CM.13 00 00 Effective Date: 15 February 2013 Version
INVESTIGATOR HANDBOOK
 INVESTIGATOR HANDBOOK Liberty IRB, Inc. 1450 S. Woodland Blvd., Suite 300A Deland, Florida 32720 Phone (386) 279-4318 Fax: (386)868-4563 Website: www.libertyirb.com Business hours: Monday Friday, 8:00am
INVESTIGATOR HANDBOOK Liberty IRB, Inc. 1450 S. Woodland Blvd., Suite 300A Deland, Florida 32720 Phone (386) 279-4318 Fax: (386)868-4563 Website: www.libertyirb.com Business hours: Monday Friday, 8:00am
Breast Cancer Registry of Greater Cincinnati (BCRGC) APPLICATION for ACCESS to INFORMATION/DATA
 APPLICATION for ACCESS to INFORMATION/DATA") Breast Cancer Registry of Greater Cincinnati (BCRGC) APPLICATION for ACCESS to INFORMATION/DATA Application Date: / / 20 Applicant's Name: LAST FIRST MIDDLE INITIAL Study Title: Institutional Affiliation(s)
Breast Cancer Registry of Greater Cincinnati (BCRGC) APPLICATION for ACCESS to INFORMATION/DATA Application Date: / / 20 Applicant's Name: LAST FIRST MIDDLE INITIAL Study Title: Institutional Affiliation(s)
Managing Risk in Clinical Research. Dr Martha J Wrigley R&D Manager Senior Visiting Fellow University of Surrey
 Managing Risk in Clinical Research Dr Martha J Wrigley R&D Manager Senior Visiting Fellow University of Surrey Aim of the session To explore the risks associated with clinical research and understand how
Managing Risk in Clinical Research Dr Martha J Wrigley R&D Manager Senior Visiting Fellow University of Surrey Aim of the session To explore the risks associated with clinical research and understand how
Reliance Agreement for Institutions Utilizing Stony Brook University s Institutional Review Board(s)
") Name of Organization Providing IRB Review: Stony Brook University ( SBU IRB ) Name of Institution Relying on the SBU IRB ( Institution ): Latest AAHRPP Accreditation Date (if applicable) OHRP Federal Wide
Name of Organization Providing IRB Review: Stony Brook University ( SBU IRB ) Name of Institution Relying on the SBU IRB ( Institution ): Latest AAHRPP Accreditation Date (if applicable) OHRP Federal Wide
UNIVERSITY OF CALIFORNIA, SAN DIEGO HUMAN RESEARCH PROTECTIONS PROGRAM. DoD/DON-funded Research
 UNIVERSITY OF CALIFORNIA, SAN DIEGO HUMAN RESEARCH PROTECTIONS PROGRAM DoD/DON-funded Research Policy In 2006, the Department of the Navy (DON) enhanced its human subject protection requirements, including
UNIVERSITY OF CALIFORNIA, SAN DIEGO HUMAN RESEARCH PROTECTIONS PROGRAM DoD/DON-funded Research Policy In 2006, the Department of the Navy (DON) enhanced its human subject protection requirements, including
Data Management & Case Report Form Development in Clinical Trials. Introduction to the Principles and Practice of Clinical Research.
 Data Management & Case Report Form Development in Clinical Trials Introduction to the Principles and Practice of Clinical Research February 3, 2015 Marge Good, RN, MPH, OCN Nurse Consultant Division of
Data Management & Case Report Form Development in Clinical Trials Introduction to the Principles and Practice of Clinical Research February 3, 2015 Marge Good, RN, MPH, OCN Nurse Consultant Division of
Electronic Medical Records and Source Data for Research: What s the Difference?
 Electronic Medical Records and Source Data for Research: What s the Difference? Tammy Anderson, CCRC, CCRA, CRCP Director, Clinical Trials Office Virginia Commonwealth University Research Coordinator 4
Electronic Medical Records and Source Data for Research: What s the Difference? Tammy Anderson, CCRC, CCRA, CRCP Director, Clinical Trials Office Virginia Commonwealth University Research Coordinator 4
This policy applies to research administrators, investigators, research staff, Human Investigation Committee (HIC) members and HIC staff.
 members and HIC staff.") REVIEW BY AN EXTERNAL INSTITUTIONAL REVIEW BOARD 233 1 of 5 PURPOSE The purpose of this policy is to establish a procedure for requesting authorization for an approved, external (non-beaumont) institutional
REVIEW BY AN EXTERNAL INSTITUTIONAL REVIEW BOARD 233 1 of 5 PURPOSE The purpose of this policy is to establish a procedure for requesting authorization for an approved, external (non-beaumont) institutional
Standard Operating Procedure on Training Requirements for staff participating in CTIMPs Sponsored by UCL
 Page 1 of 10 Standard Operating Procedure on Training Requirements for staff participating in CTIMPs Sponsored by UCL SOP ID Number: Effective Date:01/08/2012 Version Number & Date of Authorisation: V02,
Page 1 of 10 Standard Operating Procedure on Training Requirements for staff participating in CTIMPs Sponsored by UCL SOP ID Number: Effective Date:01/08/2012 Version Number & Date of Authorisation: V02,
Catherine Jahrsdorfer, RN, BSN Director of Clinical Services USF Health Office of Clinical Research
 A Practical Guide on Applying USF HRPP P&Ps in the Clinical Setting Catherine Jahrsdorfer, RN, BSN Director of Clinical Services USF Health Office of Clinical Research Learning Objectives Recognize the
A Practical Guide on Applying USF HRPP P&Ps in the Clinical Setting Catherine Jahrsdorfer, RN, BSN Director of Clinical Services USF Health Office of Clinical Research Learning Objectives Recognize the
NEGOTIATING CLINICAL TRIAL BUDGETS. Debbie Williams R.N., C.C.R.C., C.R.A., A.B.N.
 NEGOTIATING CLINICAL TRIAL BUDGETS Debbie Williams R.N., C.C.R.C., C.R.A., A.B.N. A.B.N. Amateur Budget Negotiator OVERVIEW Budget Types Analyzing the Protocol Negotiating the Contract Discussion / Tips
NEGOTIATING CLINICAL TRIAL BUDGETS Debbie Williams R.N., C.C.R.C., C.R.A., A.B.N. A.B.N. Amateur Budget Negotiator OVERVIEW Budget Types Analyzing the Protocol Negotiating the Contract Discussion / Tips
New Investigator Collaborations and Interactions: Regulatory
 Your Health and Safety... Our priority Votre santé et votre Securité notre priorité New Investigator Collaborations and Interactions: Regulatory NCIC Clinical Trials Group New Investigator Clinical Trials
Your Health and Safety... Our priority Votre santé et votre Securité notre priorité New Investigator Collaborations and Interactions: Regulatory NCIC Clinical Trials Group New Investigator Clinical Trials
Data Management in Clinical Trials
 Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research February 19, 2013 Diane St. Germain, RN, MS Nurse Consultant Division of Cancer Prevention National Cancer
Data Management in Clinical Trials Introduction to the Principles and Practice of Clinical Research February 19, 2013 Diane St. Germain, RN, MS Nurse Consultant Division of Cancer Prevention National Cancer
Conduct of clinical Trials Communication of
 PrinciPles on Conduct of clinical Trials Communication of clinical Trial results Table of Contents Preamble...1 Commitment to Protecting Research Participants...5 Conduct of Clinical Trials...7 Ensuring
PrinciPles on Conduct of clinical Trials Communication of clinical Trial results Table of Contents Preamble...1 Commitment to Protecting Research Participants...5 Conduct of Clinical Trials...7 Ensuring
Good Documentation Practices
 Good Documentation Practices Clinical Research Operations & Regulatory Support Ann Glasse, RN, BSN, MBA Director, Regulatory Support Author: Johanna Stamates, RN, MA, CCRC, CHRC Objectives Recognize the
Good Documentation Practices Clinical Research Operations & Regulatory Support Ann Glasse, RN, BSN, MBA Director, Regulatory Support Author: Johanna Stamates, RN, MA, CCRC, CHRC Objectives Recognize the
Essential Documentation and the Creation and Maintenance of Trial Master Files
 This is a controlled document. The master document is posted on the JRCO website and any print-off of this document will be classed as uncontrolled. Researchers and their teams may print off this document
This is a controlled document. The master document is posted on the JRCO website and any print-off of this document will be classed as uncontrolled. Researchers and their teams may print off this document
Guidance for IRBs, Clinical Investigators, and Sponsors
 Guidance for IRBs, Clinical Investigators, and Sponsors IRB Continuing Review after Clinical Investigation Approval U.S. Department of Health and Human Services Food and Drug Administration Center for
Guidance for IRBs, Clinical Investigators, and Sponsors IRB Continuing Review after Clinical Investigation Approval U.S. Department of Health and Human Services Food and Drug Administration Center for
Archiving of Research Documentation
 Suspension, Termination & Completion Standard Operating Procedure VERSION / REVISION: 2.0 EFFECTIVE DATE: 28 05 12 REVIEW DATE: 28 05 14 AUTHOR(S): CONTROLLER: APPROVED BY: Clinical Trials Manager; Recruitment
Suspension, Termination & Completion Standard Operating Procedure VERSION / REVISION: 2.0 EFFECTIVE DATE: 28 05 12 REVIEW DATE: 28 05 14 AUTHOR(S): CONTROLLER: APPROVED BY: Clinical Trials Manager; Recruitment
This unit will provide the structure needed to achieve successful supervision and oversight of the study. We will explain the importance of an
 Music No audio Please review information shown to understand the general course navigation and available resources. Resources, such as the audio script, example forms, and website links, can be accessed
Music No audio Please review information shown to understand the general course navigation and available resources. Resources, such as the audio script, example forms, and website links, can be accessed
Institutional Review Board
 Institutional Review Board Ethical Principles of Informed Consent Informed Consent Guidelines The principle of respect for persons requires that people be given the opportunity to choose what will or will
Institutional Review Board Ethical Principles of Informed Consent Informed Consent Guidelines The principle of respect for persons requires that people be given the opportunity to choose what will or will
Guidance for Clinical Investigators, Industry, and FDA Staff Financial Disclosure by Clinical Investigators
 Guidance for Clinical Investigators, Industry, and FDA Staff Financial Disclosure by Clinical Investigators U.S. Department of Health and Human Services Food and Drug Administration Office of Good Clinical
Guidance for Clinical Investigators, Industry, and FDA Staff Financial Disclosure by Clinical Investigators U.S. Department of Health and Human Services Food and Drug Administration Office of Good Clinical
Guidance on Withdrawal of Subjects from Research: Data Retention and Other Related Issues
 Office for Human Research Protections (OHRP) Department of Health and Human Services (HHS) Guidance on Withdrawal of Subjects from Research: Data Retention and Other Related Issues This guidance represents
Office for Human Research Protections (OHRP) Department of Health and Human Services (HHS) Guidance on Withdrawal of Subjects from Research: Data Retention and Other Related Issues This guidance represents
Introduction 2. 1. The Role of Pharmacy Within a NHS Trust 3. 2. Pharmacy Staff 4. 3. Pharmacy Facilities 5. 4. Pharmacy and Resources 6
 Index Index Section Page Introduction 2 1. The Role of Pharmacy Within a NHS Trust 3 2. Pharmacy Staff 4 3. Pharmacy Facilities 5 4. Pharmacy and Resources 6 5. Prescription Charges 7 6. Communication
Index Index Section Page Introduction 2 1. The Role of Pharmacy Within a NHS Trust 3 2. Pharmacy Staff 4 3. Pharmacy Facilities 5 4. Pharmacy and Resources 6 5. Prescription Charges 7 6. Communication
The New EU Clinical Trial Regulation Potential Impacts on Sites
 The New EU Clinical Trial Regulation Potential Impacts on Sites Angela Papa Associate Director, Clinical Management PPD Pierre-Frédéric Omnes Director, Site Start-Up and Regulatory INC Research Faculty
The New EU Clinical Trial Regulation Potential Impacts on Sites Angela Papa Associate Director, Clinical Management PPD Pierre-Frédéric Omnes Director, Site Start-Up and Regulatory INC Research Faculty
TEMPLATE DATA MANAGEMENT PLAN
 TEMPLATE DATA MANAGEMENT PLAN ICRIN (QM sub group) Version: XX Date: XXXXXXX Page 1 of 6 1.0 Document Ownership The Data Management Plan (DMP) will be initiated and subsequently owned by the Data Manager
TEMPLATE DATA MANAGEMENT PLAN ICRIN (QM sub group) Version: XX Date: XXXXXXX Page 1 of 6 1.0 Document Ownership The Data Management Plan (DMP) will be initiated and subsequently owned by the Data Manager
Department of Health and Human Services. Final Guidance Document
 Department of Health and Human Services Final Guidance Document Financial Relationships and Interests in Research Involving Human Subjects: Guidance for Human Subject Protection This document replaces
Department of Health and Human Services Final Guidance Document Financial Relationships and Interests in Research Involving Human Subjects: Guidance for Human Subject Protection This document replaces
Joint Research Office
 Office Location: 1 st Floor Maple House 149 Tottenham Court Road London W1T 7DN Joint Research Office Postal Address: UCL, Gower Street London WC1E 6BT Email: [email protected] Tel No. 020
Office Location: 1 st Floor Maple House 149 Tottenham Court Road London W1T 7DN Joint Research Office Postal Address: UCL, Gower Street London WC1E 6BT Email: [email protected] Tel No. 020
IRB Submissions and Human Subjects Research Compliance. Georgia Health Sciences University
 IRB Submissions and Human Subjects Research Compliance Offi f H R hp t ti Office of Human Research Protection Georgia Health Sciences University Objectives Identify the steps required to submit a protocol
IRB Submissions and Human Subjects Research Compliance Offi f H R hp t ti Office of Human Research Protection Georgia Health Sciences University Objectives Identify the steps required to submit a protocol
Children s Research Management System (CRMS) Version 3.0. Children s Hospital Colorado Research Institute Training Guide April 2015
 Version 3.0. Children s Hospital Colorado Research Institute Training Guide April 2015") Children s Research Management System (CRMS) Version 3.0 Children s Hospital Colorado Research Institute Training Guide April 2015 Table of Contents Operational Needs Assessment (ONA) 3 Visit Schedules
Children s Research Management System (CRMS) Version 3.0 Children s Hospital Colorado Research Institute Training Guide April 2015 Table of Contents Operational Needs Assessment (ONA) 3 Visit Schedules
Establishment of a Quality Systems approach to Clinical Site Management An Overview of Standard Operating Procedures (SOPs)
") Establishment of a Quality Systems approach to Clinical Site Management An Overview of Standard Operating Procedures (SOPs) Cornelia Kamp, MBA Executive Director Strategic Initiatives Tim Hackett Director
Establishment of a Quality Systems approach to Clinical Site Management An Overview of Standard Operating Procedures (SOPs) Cornelia Kamp, MBA Executive Director Strategic Initiatives Tim Hackett Director