Protein MW determination and protein identification by mass spectrometry
|
|
|
- Horace Robertson
- 7 years ago
- Views:
Transcription
1 Protein MW determination and protein identification by mass spectrometry Tuula Nyman Protein Chemistry Research Group Institute of Biotechnology Protein MW determination by MS (NOT= identification) -for MW determination the protein needs to be in solution without salts and detergents -usually proteins are first purified with RP chromatography before MW measurement 1
2 -MALDI TOF MS, linear mode -accuracy is not as good with ESI MS Singly charged ion Doubly charged ion Protein MW determination, ESI MS p p Protein is observed as a series of different charges -Protein MW can be calculated based on adjacent m/z-values
3 Protein MW calculation from ESI spectra p=m/z p 1 =(M r +z 1 )/z 1 p 2 =[M r +(z 1-1)]/(z 1-1) p= a peak in the mass spectrum m= total mass of an ion z= total charge M r = average mass of the protein If p 1 = and p 2 = p=m/z p 1 =(M r +z 1 )/z 1 p 2 =[M r +(z 1-1)]/(z 1-1) =(M r +z 1 )/z z 1 = Mr+z z 1 = M r = ( z 1 +z 1-1)/z z = z 1-1 ( )z 1 = z 1 = => z 1 = M r = z 1 = Da 3
 Protein identification methods: Peptide mass fingerprinting (PMF) Identification based on MS/MS data from one or more peptides")
4 Deconvoluted electrospray mass spectrum of myoglobin Protein identification by mass spectrometry protein of interest is cleaved into peptides with a specific enzyme peptides are analyzed by MS (and MS/MS) Protein identification methods: Peptide mass fingerprinting (PMF) Identification based on MS/MS data from one or more peptides 4
5 Peptide mass fingerprint (PMF) A mass spectrum of the peptide mixture resulting from the digestion of a protein by an enzyme, usually measured by MALDI-TOF Identification based on peptide MW information only Database search engines create theoretical PMFs for all the proteins in the database and compare these to the measured PMF from protein X Protein identification based on MS/MS data from one or more peptides Database search engines create theoretical peptide fragmentation patterns for all the proteins in the database and compare these to the measured MS/MS data 5
6 Protein identification by MS Protein has to be digested into peptides Disulphide bridges need to be reduced and alkylated before digestion intact protein alkylated protein tryptic fragments from the protein N S S C Reduction Alkylation N C Trypsin K R K K K K R R Protein digestion into peptides before MS analysis Digestion can be done in-solution or in-gel The enzyme has to be as specific as possible Trypsin is most commonly used enzyme: -very specific, quite cheap -cleaves peptide bond after lysines and arginines -tryptic peptides are good for MS analysis because they end up with basic amino acid -works for both in-solution and in-gel digestion 6

7 Other enzymes: -LysC, cleaves after lysines -LysN, cleaves before lysines -AspN, cleaves before aspartic acid residues -V8 protease, cleaves peptide bonds exclusively on the carbonyl side of aspartate and glutamate residues -possible to do double-digestions Chemical cleavage: Cyanogen bromide, cuts after methionines In-gel digestion -works with low-femtomolar amounts of proteins BUT: -easy to contaminate samples with keratin -the gel staining protocol needs to be compatible with MS -silver-staining: fixing with glutaraldehy cross-links protein into gel matrix after which identification is not possible 7
8 Peptide mass fingerprinting -usually peptides need to be desalted and concentrated before MALDI analysis -> ZipTips, peptide elution directly onto MALDI target plate -MALDI matrix included in the elution solution Peptide mass fingerprinting MALDI, Positive ion reflector mode -silver-stained spots from 2-DE gels, in-gel digestion+ziptipping 8
![5 4 3 2 1 1574.775 0 1568 15 70 1572 15 74 15 76 1578 1580 1582 m /z Intens. [a.u.] x10 4 Peak picking parameters important in PMF Intens. [a.u.] x10 4 1590.760 1574.](/docs-images/58/41670530/images/9-0.png "775 5 Label monoisotopic masses Check for correct isotope! 4 1434.642 3 2 1 1606.761 1450.637 1475.728 1657.")
9 m /z Intens. [a.u.] x10 4 Peak picking parameters important in PMF Intens. [a.u.] x Label monoisotopic masses Check for correct isotope! m/z Publicly available search engines for PMF Mascot ProteinProspector/ MS-Fit PROWL/ ProFound Aldente Databases 9
10 PMF/ database search 10
11 11
 -possibility to do 2nd pass search Normal search, max 1missed cleavage allowed Max 4 missed cleavages allowed")
12 Usually not many missed cleavages Mass accuracy critical -check for known contaminants (these should be removed before search) -possibility to do 2nd pass search Normal search, max 1missed cleavage allowed Max 4 missed cleavages allowed 12
13 PEPTIDE MASS FINGERPRINTING quick and easy requires a very spesific enzyme optimized digestion+ desalting protocols internal/close external calibration of MALDI spectra works only for proteins which are already in the databases as protein sequences not suitable for complex protein mixtures Protein identification based on MS/MS data from one or more peptides -MALDI-TOF/TOF or nanolc-esi-ms/ms analysis -suitable for (complex) protein mixtures, especially when combined with LC separation of peptides before MS/MS 13
14 Tandem mass spectrometry scan types Product ion scanning MS scan: The ions are first separated according to their m/z ratios MS/MS scan: Peptides with certain m/z-ratio are selected and fragmented inside the mass analyzer, and the m/z-ratios of the fragment ions are measured Intensity, counts TOF product 745,4 C H I L L P L I H m/z, amu 14
15 Peptide Fragmentation Nomenclature Peptides do not fragment sequentially, the fragmentation events are somewhat random. The most common peptide fragments observed in low energy collisions are a, b and y ions. The b ions appear to extend from the amino terminus (N-terminus), and y ions appear to extend from the carboxyl terminus (C-terminus). Protein identification with MALDI-TOF/TOF -first, PMF with MALDI-TOF -next, selected precursor ions can be fragmented (TOF/TOF analysis) -MALDI produces singly charged parent ions product ion spectra not as easy to interpret as in ESI-MS/MS -database search with both PMF and MS/MS information 15

16 m/z m /z MALDI-TOF/TOF fragment ion spectra from parent ion m/z 1952 Intens. [a.u.] x Intens. [a.u.] x Sequence Query: One or more peptide mass values associated with information such as partial or ambiguous sequence strings, amino acid composition information, MS/MS fragment ion masses, etc. 16

17 Protein identification using nanolc-ms/ms um i.d. RP-columns, 200 nl/min no need to split the effluent before MS -DDA= data dependent analysis, can be fully automated -suitable for complex protein mixtures, possibility to identify hundreds of proteins in one run 17
18 DDA =Data Dependent Acquisition (IDA =Information Dependent Acquisition) -fully automated experiment, first MS scan followed by two or more product ion scans -the acquisition software is set to choose certain types of ions for fragmentation and to use suitable collision energy for this ion -in ESI tryptic peptides have usually 2-4 charges DDA experiment Exp 1 = TOF MS Exp 2 = Product ion scan from parent I Exp 3 = Product ion scan from parent II 18
19 TIC = total ion current Exp 1 = TOF MS Exp 2 = Product ion scan I Exp 3 = Product ion scan II 19
20 Search engines for (LC-)MS/MS data -Mascot, Sequest, OMSSA etc -the programs take the fragment ion spectrum of a peptide as input and score it against theoretical fragmentation patterns constructed for peptides from the searched database. -in practise the user is often limited to use those search engines which accept the data format from the mass spec used -mzxml is a open data format for storage and exchange of mass spec data -raw, proprietary file formats from most vendors can be converted to the open mzxml format 20

21 Ion scores from individual MS/MS spectra Protein identification from complex mixtures using nanolc-ms/ms -produces huge amounts of raw data -requires efficient data processing tools -different database search programs can produce different results from the same raw data -false discovery rate estimation 21
22 Comparative proteome cataloging of Lactobacillus rhamnosus strains GG and Lc705 -in-depth proteome analysis of two Lactobacillus rhamnosus strains, the well-known probiotic strain GG and the dairy strain Lc705 -GeLC-MS/MS: proteins are separated using SDS_PAGE and identified using nanolc-ms/ms -to maximize the number of identifications, all data sets were searched against the target databases using two search engines, Mascot and Paragon Savijoki et al, JPR
23 Data analyses Database Searches Protein Pilot (Mascot & Paragon) Compid analyses Compilation of Mascot and Paragon results Bioinformatics Cellular location of the identified proteins Savijoki et al, JPR 2011 Natri et al, JPR
24 Comparison of protein ID results from the same raw data with different search engines ProICAT SP2 Spectrum Mill Sequest Moulder et al, Proteomics. 2005;5: Comparison of the ID results with different search engines a)peptide identifications, b) Proteins identifications with two or more peptides given as a proportion of all protein identifications. 24
25 False discovery rate (FDR) estimation FDR = FP/(TP+FP) TP= true positive matches FP =false positive matches Previously: search the raw data first against the normal database and then against the decoy database -> calculate FDR based on these NOW the preferred method: One search against a database in which the target and decoy sequences have been concatenated. This means that you will only record a false positive when a match from the decoy sequences is better than any match from the target sequences. Elias, J. E., et al, Nature Methods (2005). Now you re protein is identified What else can we find in the data? Protein fragmentation -protein separation by SDS-PAGE/2-DE followed by in-gel digestion and MS analysis 25

26 Cytokeratin-18 is cleaved during viral infection, and fragments localize onto mitochondria Mitochondrial protemes Based on 2-DE and MS data: N-terminal fragment of the identified protein 26

27 Post-translational modifications TAP-tag purification of protein complexes, on-beads digestion with double enzyme, first LysC 2h, then trypsin o/n 27
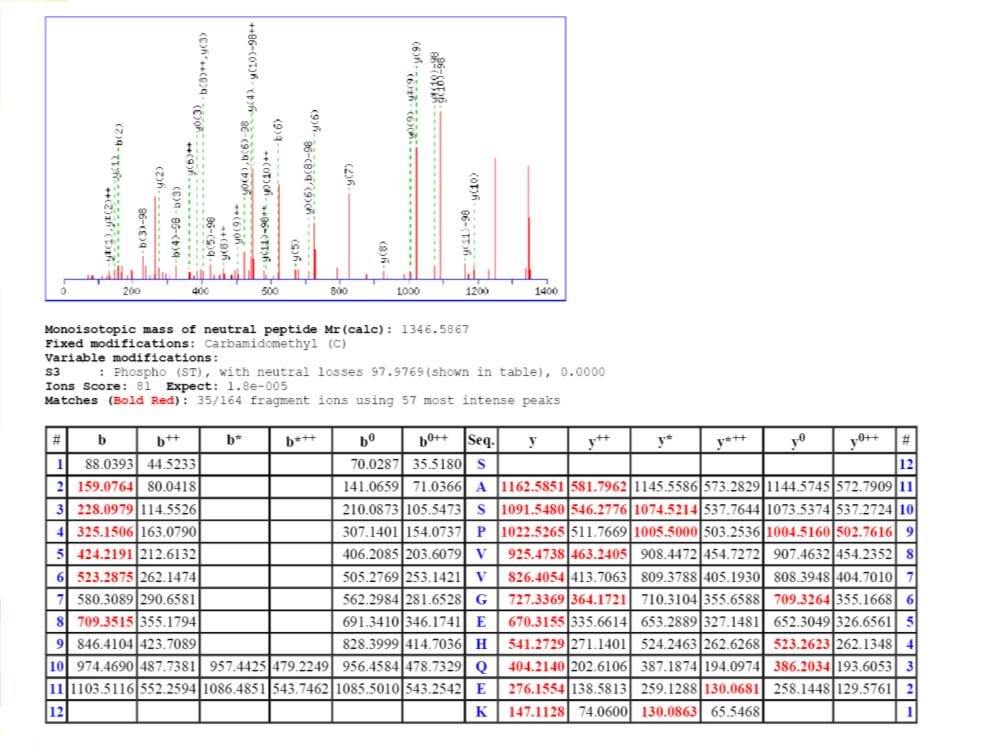
28 KIN2 phosphopeptide 28
29 De novo sequencing De novo =peptide sequencing performed without prior knowledge of the amino acid sequence -if enough material is available classical Edman degradation is still a vey good method -partial peptide sequencing is possible based on MS/MS data 29
Master course KEMM03 Principles of Mass Spectrometric Protein Characterization. Exam
 Exam Master course KEMM03 Principles of Mass Spectrometric Protein Characterization 2010-10-29 kl 08.15-13.00 Use a new paper for answering each question! Write your name on each paper! Aids: Mini calculator,
Exam Master course KEMM03 Principles of Mass Spectrometric Protein Characterization 2010-10-29 kl 08.15-13.00 Use a new paper for answering each question! Write your name on each paper! Aids: Mini calculator,
Laboration 1. Identifiering av proteiner med Mass Spektrometri. Klinisk Kemisk Diagnostik
 Laboration 1 Identifiering av proteiner med Mass Spektrometri Klinisk Kemisk Diagnostik Sven Kjellström 2014 kjellstrom.sven@gmail.com 0702-935060 Laboration 1 Klinisk Kemisk Diagnostik Identifiering av
Laboration 1 Identifiering av proteiner med Mass Spektrometri Klinisk Kemisk Diagnostik Sven Kjellström 2014 kjellstrom.sven@gmail.com 0702-935060 Laboration 1 Klinisk Kemisk Diagnostik Identifiering av
In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates
 In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates Using the Explore Workflow on the AB SCIEX TripleTOF 5600 System A major challenge in proteomics
In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates Using the Explore Workflow on the AB SCIEX TripleTOF 5600 System A major challenge in proteomics
AB SCIEX TOF/TOF 4800 PLUS SYSTEM. Cost effective flexibility for your core needs
 AB SCIEX TOF/TOF 4800 PLUS SYSTEM Cost effective flexibility for your core needs AB SCIEX TOF/TOF 4800 PLUS SYSTEM It s just what you expect from the industry leader. The AB SCIEX 4800 Plus MALDI TOF/TOF
AB SCIEX TOF/TOF 4800 PLUS SYSTEM Cost effective flexibility for your core needs AB SCIEX TOF/TOF 4800 PLUS SYSTEM It s just what you expect from the industry leader. The AB SCIEX 4800 Plus MALDI TOF/TOF
Effects of Intelligent Data Acquisition and Fast Laser Speed on Analysis of Complex Protein Digests
 Effects of Intelligent Data Acquisition and Fast Laser Speed on Analysis of Complex Protein Digests AB SCIEX TOF/TOF 5800 System with DynamicExit Algorithm and ProteinPilot Software for Robust Protein
Effects of Intelligent Data Acquisition and Fast Laser Speed on Analysis of Complex Protein Digests AB SCIEX TOF/TOF 5800 System with DynamicExit Algorithm and ProteinPilot Software for Robust Protein
ProteinScape. Innovation with Integrity. Proteomics Data Analysis & Management. Mass Spectrometry
 ProteinScape Proteomics Data Analysis & Management Innovation with Integrity Mass Spectrometry ProteinScape a Virtual Environment for Successful Proteomics To overcome the growing complexity of proteomics
ProteinScape Proteomics Data Analysis & Management Innovation with Integrity Mass Spectrometry ProteinScape a Virtual Environment for Successful Proteomics To overcome the growing complexity of proteomics
Tutorial for Proteomics Data Submission. Katalin F. Medzihradszky Robert J. Chalkley UCSF
 Tutorial for Proteomics Data Submission Katalin F. Medzihradszky Robert J. Chalkley UCSF Why Have Guidelines? Large-scale proteomics studies create huge amounts of data. It is impossible/impractical to
Tutorial for Proteomics Data Submission Katalin F. Medzihradszky Robert J. Chalkley UCSF Why Have Guidelines? Large-scale proteomics studies create huge amounts of data. It is impossible/impractical to
Application Note # LCMS-81 Introducing New Proteomics Acquisiton Strategies with the compact Towards the Universal Proteomics Acquisition Method
 Application Note # LCMS-81 Introducing New Proteomics Acquisiton Strategies with the compact Towards the Universal Proteomics Acquisition Method Introduction During the last decade, the complexity of samples
Application Note # LCMS-81 Introducing New Proteomics Acquisiton Strategies with the compact Towards the Universal Proteomics Acquisition Method Introduction During the last decade, the complexity of samples
Aiping Lu. Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn
 Aiping Lu Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn Proteome and Proteomics PROTEin complement expressed by genome Marc Wilkins Electrophoresis. 1995. 16(7):1090-4. proteomics
Aiping Lu Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn Proteome and Proteomics PROTEin complement expressed by genome Marc Wilkins Electrophoresis. 1995. 16(7):1090-4. proteomics
Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance?
 Optimization 1 Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance? Where to begin? 2 Sequence Databases Swiss-prot MSDB, NCBI nr dbest Species specific ORFS
Optimization 1 Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance? Where to begin? 2 Sequence Databases Swiss-prot MSDB, NCBI nr dbest Species specific ORFS
ProteinPilot Report for ProteinPilot Software
 ProteinPilot Report for ProteinPilot Software Detailed Analysis of Protein Identification / Quantitation Results Automatically Sean L Seymour, Christie Hunter SCIEX, USA Pow erful mass spectrometers like
ProteinPilot Report for ProteinPilot Software Detailed Analysis of Protein Identification / Quantitation Results Automatically Sean L Seymour, Christie Hunter SCIEX, USA Pow erful mass spectrometers like
Application Note # MT-90 MALDI-TDS: A Coherent MALDI Top-Down-Sequencing Approach Applied to the ABRF-Protein Research Group Study 2008
 Bruker Daltonics Application Note # MT-90 MALDI-TDS: A Coherent MALDI Top-Down-Sequencing Approach Applied to the ABRF-Protein Research Group Study 2008 In the ABRF-PRG study 2008 [*] the ability to characterize
Bruker Daltonics Application Note # MT-90 MALDI-TDS: A Coherent MALDI Top-Down-Sequencing Approach Applied to the ABRF-Protein Research Group Study 2008 In the ABRF-PRG study 2008 [*] the ability to characterize
Introduction to Database Searching using MASCOT
 Introduction to Database Searching using MASCOT 1 Three ways to use mass spectrometry data for protein identification 1.Peptide Mass Fingerprint A set of peptide molecular masses from an enzyme digest
Introduction to Database Searching using MASCOT 1 Three ways to use mass spectrometry data for protein identification 1.Peptide Mass Fingerprint A set of peptide molecular masses from an enzyme digest
LABORATÓRIUMI GYAKORLAT SILLABUSZ SYLLABUS OF A PRACTICAL DEMONSTRATION. financed by the program
 TÁMOP-4.1.1.C-13/1/KONV-2014-0001 projekt Az élettudományi-klinikai felsőoktatás gyakorlatorientált és hallgatóbarát korszerűsítése a vidéki képzőhelyek nemzetközi versenyképességének erősítésére program
TÁMOP-4.1.1.C-13/1/KONV-2014-0001 projekt Az élettudományi-klinikai felsőoktatás gyakorlatorientált és hallgatóbarát korszerűsítése a vidéki képzőhelyek nemzetközi versenyképességének erősítésére program
Proteomics in Practice
 Reiner Westermeier, Torn Naven Hans-Rudolf Höpker Proteomics in Practice A Guide to Successful Experimental Design 2008 Wiley-VCH Verlag- Weinheim 978-3-527-31941-1 Preface Foreword XI XIII Abbreviations,
Reiner Westermeier, Torn Naven Hans-Rudolf Höpker Proteomics in Practice A Guide to Successful Experimental Design 2008 Wiley-VCH Verlag- Weinheim 978-3-527-31941-1 Preface Foreword XI XIII Abbreviations,
PROTEIN SEQUENCING. First Sequence
 PROTEIN SEQUENCING First Sequence The first protein sequencing was achieved by Frederic Sanger in 1953. He determined the amino acid sequence of bovine insulin Sanger was awarded the Nobel Prize in 1958
PROTEIN SEQUENCING First Sequence The first protein sequencing was achieved by Frederic Sanger in 1953. He determined the amino acid sequence of bovine insulin Sanger was awarded the Nobel Prize in 1958
Mass Spectra Alignments and their Significance
 Mass Spectra Alignments and their Significance Sebastian Böcker 1, Hans-Michael altenbach 2 1 Technische Fakultät, Universität Bielefeld 2 NRW Int l Graduate School in Bioinformatics and Genome Research,
Mass Spectra Alignments and their Significance Sebastian Böcker 1, Hans-Michael altenbach 2 1 Technische Fakultät, Universität Bielefeld 2 NRW Int l Graduate School in Bioinformatics and Genome Research,
Increasing the Multiplexing of High Resolution Targeted Peptide Quantification Assays
 Increasing the Multiplexing of High Resolution Targeted Peptide Quantification Assays Scheduled MRM HR Workflow on the TripleTOF Systems Jenny Albanese, Christie Hunter AB SCIEX, USA Targeted quantitative
Increasing the Multiplexing of High Resolution Targeted Peptide Quantification Assays Scheduled MRM HR Workflow on the TripleTOF Systems Jenny Albanese, Christie Hunter AB SCIEX, USA Targeted quantitative
Definition of the Measurand: CRP
 A Reference Measurement System for C-reactive Protein David M. Bunk, Ph.D. Chemical Science and Technology Laboratory National Institute of Standards and Technology Definition of the Measurand: Human C-reactive
A Reference Measurement System for C-reactive Protein David M. Bunk, Ph.D. Chemical Science and Technology Laboratory National Institute of Standards and Technology Definition of the Measurand: Human C-reactive
Workshop IIc. Manual interpretation of MS/MS spectra. Ebbing de Jong. Center for Mass Spectrometry and Proteomics Phone (612)625-2280 (612)625-2279
625-2280 (612)625-2279") Workshop IIc Manual interpretation of MS/MS spectra Ebbing de Jong Why MS/MS spectra? The information contained in an MS spectrum (m/z, isotope spacing and therefore z ) is not enough to tell us the amino
Workshop IIc Manual interpretation of MS/MS spectra Ebbing de Jong Why MS/MS spectra? The information contained in an MS spectrum (m/z, isotope spacing and therefore z ) is not enough to tell us the amino
Mass Spectrometry Based Proteomics
 Mass Spectrometry Based Proteomics Proteomics Shared Research Oregon Health & Science University Portland, Oregon This document is designed to give a brief overview of Mass Spectrometry Based Proteomics
Mass Spectrometry Based Proteomics Proteomics Shared Research Oregon Health & Science University Portland, Oregon This document is designed to give a brief overview of Mass Spectrometry Based Proteomics
Global and Discovery Proteomics Lecture Agenda
 Global and Discovery Proteomics Christine A. Jelinek, Ph.D. Johns Hopkins University School of Medicine Department of Pharmacology and Molecular Sciences Middle Atlantic Mass Spectrometry Laboratory Global
Global and Discovery Proteomics Christine A. Jelinek, Ph.D. Johns Hopkins University School of Medicine Department of Pharmacology and Molecular Sciences Middle Atlantic Mass Spectrometry Laboratory Global
Error Tolerant Searching of Uninterpreted MS/MS Data
 Error Tolerant Searching of Uninterpreted MS/MS Data 1 In any search of a large LC-MS/MS dataset 2 There are always a number of spectra which get poor scores, or even no match at all. 3 Sometimes, this
Error Tolerant Searching of Uninterpreted MS/MS Data 1 In any search of a large LC-MS/MS dataset 2 There are always a number of spectra which get poor scores, or even no match at all. 3 Sometimes, this
ProSightPC 3.0 Quick Start Guide
 ProSightPC 3.0 Quick Start Guide The Thermo ProSightPC 3.0 application is the only proteomics software suite that effectively supports high-mass-accuracy MS/MS experiments performed on LTQ FT and LTQ Orbitrap
ProSightPC 3.0 Quick Start Guide The Thermo ProSightPC 3.0 application is the only proteomics software suite that effectively supports high-mass-accuracy MS/MS experiments performed on LTQ FT and LTQ Orbitrap
Pep-Miner: A Novel Technology for Mass Spectrometry-Based Proteomics
 Pep-Miner: A Novel Technology for Mass Spectrometry-Based Proteomics Ilan Beer Haifa Research Lab Dec 10, 2002 Pep-Miner s Location in the Life Sciences World The post-genome era - the age of proteome
Pep-Miner: A Novel Technology for Mass Spectrometry-Based Proteomics Ilan Beer Haifa Research Lab Dec 10, 2002 Pep-Miner s Location in the Life Sciences World The post-genome era - the age of proteome
MASCOT Search Results Interpretation
 The Mascot protein identification program (Matrix Science, Ltd.) uses statistical methods to assess the validity of a match. MS/MS data is not ideal. That is, there are unassignable peaks (noise) and usually
The Mascot protein identification program (Matrix Science, Ltd.) uses statistical methods to assess the validity of a match. MS/MS data is not ideal. That is, there are unassignable peaks (noise) and usually
Session 1. Course Presentation: Mass spectrometry-based proteomics for molecular and cellular biologists
 Program Overview Session 1. Course Presentation: Mass spectrometry-based proteomics for molecular and cellular biologists Session 2. Principles of Mass Spectrometry Session 3. Mass spectrometry based proteomics
Program Overview Session 1. Course Presentation: Mass spectrometry-based proteomics for molecular and cellular biologists Session 2. Principles of Mass Spectrometry Session 3. Mass spectrometry based proteomics
Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench
 Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench Application Guide Agilent Technologies Notices Agilent Technologies, Inc. 2012 No part of this manual may be reproduced in any form or by any
Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench Application Guide Agilent Technologies Notices Agilent Technologies, Inc. 2012 No part of this manual may be reproduced in any form or by any
Introduction to Proteomics 1.0
 Introduction to Proteomics 1.0 CMSP Workshop Tim Griffin Associate Professor, BMBB Faculty Director, CMSP Objectives Why are we here? For participants: Learn basics of MS-based proteomics Learn what s
Introduction to Proteomics 1.0 CMSP Workshop Tim Griffin Associate Professor, BMBB Faculty Director, CMSP Objectives Why are we here? For participants: Learn basics of MS-based proteomics Learn what s
SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays
 Protocol SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878 Order per fax: +49-30-6392-7888 or e-mail: www: peptide@jpt.com www.jpt.com
Protocol SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878 Order per fax: +49-30-6392-7888 or e-mail: www: peptide@jpt.com www.jpt.com
泛 用 蛋 白 質 體 學 之 質 譜 儀 資 料 分 析 平 台 的 建 立 與 應 用 Universal Mass Spectrometry Data Analysis Platform for Quantitative and Qualitative Proteomics
 泛 用 蛋 白 質 體 學 之 質 譜 儀 資 料 分 析 平 台 的 建 立 與 應 用 Universal Mass Spectrometry Data Analysis Platform for Quantitative and Qualitative Proteomics 2014 Training Course Wei-Hung Chang ( 張 瑋 宏 ) ABRC, Academia
泛 用 蛋 白 質 體 學 之 質 譜 儀 資 料 分 析 平 台 的 建 立 與 應 用 Universal Mass Spectrometry Data Analysis Platform for Quantitative and Qualitative Proteomics 2014 Training Course Wei-Hung Chang ( 張 瑋 宏 ) ABRC, Academia
Challenges in Computational Analysis of Mass Spectrometry Data for Proteomics
 Ma B. Challenges in computational analysis of mass spectrometry data for proteomics. SCIENCE AND TECHNOLOGY 25(1): 1 Jan. 2010 JOURNAL OF COMPUTER Challenges in Computational Analysis of Mass Spectrometry
Ma B. Challenges in computational analysis of mass spectrometry data for proteomics. SCIENCE AND TECHNOLOGY 25(1): 1 Jan. 2010 JOURNAL OF COMPUTER Challenges in Computational Analysis of Mass Spectrometry
Principles and Applications of Proteomics
 Principles and Applications of Proteomics Why Proteomics? 2-DE Overview Sample preparation 1 st & 2 nd dimension seperation Data Analysis Sample preparation for Mass Spectrometry Mass Spectrometry MALDI-TOF,
Principles and Applications of Proteomics Why Proteomics? 2-DE Overview Sample preparation 1 st & 2 nd dimension seperation Data Analysis Sample preparation for Mass Spectrometry Mass Spectrometry MALDI-TOF,
Introduction to Proteomics
 Introduction to Proteomics Why Proteomics? Same Genome Different Proteome Black Swallowtail - larvae and butterfly Biological Complexity Yeast - a simple proteome 6,113 proteins = 344,855 tryptic peptides
Introduction to Proteomics Why Proteomics? Same Genome Different Proteome Black Swallowtail - larvae and butterfly Biological Complexity Yeast - a simple proteome 6,113 proteins = 344,855 tryptic peptides
Standard Mixture. TOF/TOF Calibration Mixture. Calibration Mixture 1 (Cal Mix 1, 1:10) Calibration Mixture 1 (Cal Mix 1, 1:100)
 Calibration Mixture 1 (Cal Mix 1, 1:100)") Mass Standards Kit for Calibration of AB SCIEX TOF/TOF Instruments Protocol 1 Product Description The Mass Standards Kit includes reagents needed to test instrument function, optimize instrument parameters,
Mass Standards Kit for Calibration of AB SCIEX TOF/TOF Instruments Protocol 1 Product Description The Mass Standards Kit includes reagents needed to test instrument function, optimize instrument parameters,
How Mascot Integra helps run a Core Lab
 How Mascot Integra helps run a Core Lab 1 Areas where a database can help a core lab Project, experiment and sample tracking Flexibility in experiment design Role based security Automation Custom results
How Mascot Integra helps run a Core Lab 1 Areas where a database can help a core lab Project, experiment and sample tracking Flexibility in experiment design Role based security Automation Custom results
Methods for Protein Analysis
 Methods for Protein Analysis 1. Protein Separation Methods The following is a quick review of some common methods used for protein separation: SDS-PAGE (SDS-polyacrylamide gel electrophoresis) separates
Methods for Protein Analysis 1. Protein Separation Methods The following is a quick review of some common methods used for protein separation: SDS-PAGE (SDS-polyacrylamide gel electrophoresis) separates
Advantages of the LTQ Orbitrap for Protein Identification in Complex Digests
 Application Note: 386 Advantages of the LTQ Orbitrap for Protein Identification in Complex Digests Rosa Viner, Terry Zhang, Scott Peterman, and Vlad Zabrouskov, Thermo Fisher Scientific, San Jose, CA,
Application Note: 386 Advantages of the LTQ Orbitrap for Protein Identification in Complex Digests Rosa Viner, Terry Zhang, Scott Peterman, and Vlad Zabrouskov, Thermo Fisher Scientific, San Jose, CA,
MaxQuant User s Guide Version 1.2.2.5
 MaxQuant User s Guide Version 1.2.2.5 Jűrgen Cox and Matthias Mann Nature Biotechnology 26, 1367-1372 (2008) Sara ten Have 2012 http://www.lamondlab.com/ http://greproteomics.lifesci.dundee.ac.uk/ References
MaxQuant User s Guide Version 1.2.2.5 Jűrgen Cox and Matthias Mann Nature Biotechnology 26, 1367-1372 (2008) Sara ten Have 2012 http://www.lamondlab.com/ http://greproteomics.lifesci.dundee.ac.uk/ References
A. A peptide with 12 amino acids has the following amino acid composition: 2 Met, 1 Tyr, 1 Trp, 2 Glu, 1 Lys, 1 Arg, 1 Thr, 1 Asn, 1 Ile, 1 Cys
 Questions- Proteins & Enzymes A. A peptide with 12 amino acids has the following amino acid composition: 2 Met, 1 Tyr, 1 Trp, 2 Glu, 1 Lys, 1 Arg, 1 Thr, 1 Asn, 1 Ile, 1 Cys Reaction of the intact peptide
Questions- Proteins & Enzymes A. A peptide with 12 amino acids has the following amino acid composition: 2 Met, 1 Tyr, 1 Trp, 2 Glu, 1 Lys, 1 Arg, 1 Thr, 1 Asn, 1 Ile, 1 Cys Reaction of the intact peptide
(c) How would your answers to problem (a) change if the molecular weight of the protein was 100,000 Dalton?
 How would your answers to problem (a) change if the molecular weight of the protein was 100,000 Dalton?") Problem 1. (12 points total, 4 points each) The molecular weight of an unspecified protein, at physiological conditions, is 70,000 Dalton, as determined by sedimentation equilibrium measurements and by
Problem 1. (12 points total, 4 points each) The molecular weight of an unspecified protein, at physiological conditions, is 70,000 Dalton, as determined by sedimentation equilibrium measurements and by
MultiQuant Software 2.0 for Targeted Protein / Peptide Quantification
 MultiQuant Software 2.0 for Targeted Protein / Peptide Quantification Gold Standard for Quantitative Data Processing Because of the sensitivity, selectivity, speed and throughput at which MRM assays can
MultiQuant Software 2.0 for Targeted Protein / Peptide Quantification Gold Standard for Quantitative Data Processing Because of the sensitivity, selectivity, speed and throughput at which MRM assays can
Biochemistry - I. Prof. S. Dasgupta Department of Chemistry Indian Institute of Technology, Kharagpur Lecture-11 Enzyme Mechanisms II
 Biochemistry - I Prof. S. Dasgupta Department of Chemistry Indian Institute of Technology, Kharagpur Lecture-11 Enzyme Mechanisms II In the last class we studied the enzyme mechanisms of ribonuclease A
Biochemistry - I Prof. S. Dasgupta Department of Chemistry Indian Institute of Technology, Kharagpur Lecture-11 Enzyme Mechanisms II In the last class we studied the enzyme mechanisms of ribonuclease A
ANALYSIS OF PROTEINS AND PROTEOMES BY MASS SPECTROMETRY
 Annu. Rev. Biochem. 2001. 70:437 73 Copyright c 2001 by Annual Reviews. All rights reserved ANALYSIS OF PROTEINS AND PROTEOMES BY MASS SPECTROMETRY Matthias Mann, 1,2 Ronald C. Hendrickson, 2 and Akhilesh
Annu. Rev. Biochem. 2001. 70:437 73 Copyright c 2001 by Annual Reviews. All rights reserved ANALYSIS OF PROTEINS AND PROTEOMES BY MASS SPECTROMETRY Matthias Mann, 1,2 Ronald C. Hendrickson, 2 and Akhilesh
THE ABC S (AND XYZ S) OF PEPTIDE SEQUENCING
 OF PEPTIDE SEQUENCING") TE ABC S (AD XYZ S) F PEPTIDE SEQUECIG anno Steen* and Matthias Mann Abstract Proteomics is an increasingly powerful and indispensable technology in molecular cell biology. It can be used to identify the
TE ABC S (AD XYZ S) F PEPTIDE SEQUECIG anno Steen* and Matthias Mann Abstract Proteomics is an increasingly powerful and indispensable technology in molecular cell biology. It can be used to identify the
Protein Hit1, a novel box C/D snornp assembly factor, controls cellular concentration of protein Rsa1p by direct interaction
 Supplementary data Protein Hit1, a novel box C/D snornp assembly factor, controls cellular concentration of protein Rsa1p by direct interaction Benjamin Rothé, Jean-Michel Saliou, Marc Quinternet, Régis
Supplementary data Protein Hit1, a novel box C/D snornp assembly factor, controls cellular concentration of protein Rsa1p by direct interaction Benjamin Rothé, Jean-Michel Saliou, Marc Quinternet, Régis
SimGlycan Software*: A New Predictive Carbohydrate Analysis Tool for MS/MS Data
 SimGlycan Software*: A New Predictive Carbohydrate Analysis Tool for MS/MS Data Automated Data Interpretation for Glycan Characterization Jenny Albanese 1, Matthias Glueckmann 2 and Christof Lenz 2 1 AB
SimGlycan Software*: A New Predictive Carbohydrate Analysis Tool for MS/MS Data Automated Data Interpretation for Glycan Characterization Jenny Albanese 1, Matthias Glueckmann 2 and Christof Lenz 2 1 AB
Proteomic Analysis using Accurate Mass Tags. Gordon Anderson PNNL January 4-5, 2005
 Proteomic Analysis using Accurate Mass Tags Gordon Anderson PNNL January 4-5, 2005 Outline Accurate Mass and Time Tag (AMT) based proteomics Instrumentation Data analysis Data management Challenges 2 Approach
Proteomic Analysis using Accurate Mass Tags Gordon Anderson PNNL January 4-5, 2005 Outline Accurate Mass and Time Tag (AMT) based proteomics Instrumentation Data analysis Data management Challenges 2 Approach
Quantitative mass spec based proteomics
 Quantitative mass spec based proteomics Tuula Nyman Institute of Biotechnology tuula.nyman@helsinki.fi Proteomics is the large-scale study of proteins Proteomics provides information on: -protein expression
Quantitative mass spec based proteomics Tuula Nyman Institute of Biotechnology tuula.nyman@helsinki.fi Proteomics is the large-scale study of proteins Proteomics provides information on: -protein expression
Marmara Üniversitesi Fen-Edebiyat Fakültesi Kimya Bölümü / Biyokimya Anabilim Dalı
 EXPERIMENT IX Marmara Üniversitesi DETERMINATION OF N-TERMINAL AMINO ACID RESIDUE OF PROTEINS BY THIN LAYER CHROMATOGRAPHY Functions of the proteins depend upon its amino acid sequence. Because amino acid
EXPERIMENT IX Marmara Üniversitesi DETERMINATION OF N-TERMINAL AMINO ACID RESIDUE OF PROTEINS BY THIN LAYER CHROMATOGRAPHY Functions of the proteins depend upon its amino acid sequence. Because amino acid
Mass spectrometry based proteomics
 Practice-oriented, student-friendly modernization of the biomedical education for strengthening the international competitiveness of the rural Hungarian universities TÁMOP-4.1.1.C-13/1/KONV-2014-0001 Mass
Practice-oriented, student-friendly modernization of the biomedical education for strengthening the international competitiveness of the rural Hungarian universities TÁMOP-4.1.1.C-13/1/KONV-2014-0001 Mass
for mass spectrometry calibration tools Thermo Scientific Pierce Controls and Standards for Mass Spectrometry
 Thermo Scientific Pierce Controls and Standards for Mass Spectrometry calibration tools for mass spectrometry Ensure confidence in instrument performance with Thermo Scientific Pierce Calibration Solutions
Thermo Scientific Pierce Controls and Standards for Mass Spectrometry calibration tools for mass spectrometry Ensure confidence in instrument performance with Thermo Scientific Pierce Calibration Solutions
MRMPilot Software: Accelerating MRM Assay Development for Targeted Quantitative Proteomics
 MRMPilot Software: Accelerating MRM Assay Development for Targeted Quantitative Proteomics With Unique QTRAP and TripleTOF 5600 System Technology Targeted peptide quantification is a rapidly growing application
MRMPilot Software: Accelerating MRM Assay Development for Targeted Quantitative Proteomics With Unique QTRAP and TripleTOF 5600 System Technology Targeted peptide quantification is a rapidly growing application
Protein Prospector and Ways of Calculating Expectation Values
 Protein Prospector and Ways of Calculating Expectation Values 1/16 Aenoch J. Lynn; Robert J. Chalkley; Peter R. Baker; Mark R. Segal; and Alma L. Burlingame University of California, San Francisco, San
Protein Prospector and Ways of Calculating Expectation Values 1/16 Aenoch J. Lynn; Robert J. Chalkley; Peter R. Baker; Mark R. Segal; and Alma L. Burlingame University of California, San Francisco, San
Fast and Automatic Mapping of Disulfide Bonds in a Monoclonal Antibody using SYNAPT G2 HDMS and BiopharmaLynx 1.3
 Fast and Automatic Mapping of Disulfide Bonds in a Monoclonal Antibody using SYNAPT G2 HDMS and BiopharmaLynx 1.3 Hongwei Xie and Weibin Chen Waters Corporation, Milford, MA, U.S. A P P L I C AT ION B
Fast and Automatic Mapping of Disulfide Bonds in a Monoclonal Antibody using SYNAPT G2 HDMS and BiopharmaLynx 1.3 Hongwei Xie and Weibin Chen Waters Corporation, Milford, MA, U.S. A P P L I C AT ION B
Proteomic data analysis for Orbitrap datasets using Resources available at MSI. September 28 th 2011 Pratik Jagtap
 Proteomic data analysis for Orbitrap datasets using Resources available at MSI. September 28 th 2011 Pratik Jagtap The Minnesota http://www.mass.msi.umn.edu/ Proteomics workflow Trypsin Protein Peptides
Proteomic data analysis for Orbitrap datasets using Resources available at MSI. September 28 th 2011 Pratik Jagtap The Minnesota http://www.mass.msi.umn.edu/ Proteomics workflow Trypsin Protein Peptides
Electrospray Ion Trap Mass Spectrometry. Introduction
 Electrospray Ion Source Electrospray Ion Trap Mass Spectrometry Introduction The key to using MS for solutions is the ability to transfer your analytes into the vacuum of the mass spectrometer as ionic
Electrospray Ion Source Electrospray Ion Trap Mass Spectrometry Introduction The key to using MS for solutions is the ability to transfer your analytes into the vacuum of the mass spectrometer as ionic
Shotgun Proteomic Analysis. Department of Cell Biology The Scripps Research Institute
 Shotgun Proteomic Analysis Department of Cell Biology The Scripps Research Institute Biological/Functional Resolution of Experiments Organelle Multiprotein Complex Cells/Tissues Function Expression Analysis
Shotgun Proteomic Analysis Department of Cell Biology The Scripps Research Institute Biological/Functional Resolution of Experiments Organelle Multiprotein Complex Cells/Tissues Function Expression Analysis
La Protéomique : Etat de l art et perspectives
 La Protéomique : Etat de l art et perspectives Odile Schiltz Institut de Pharmacologie et de Biologie Structurale CNRS, Université de Toulouse, Odile.Schiltz@ipbs.fr Protéomique et Spectrométrie de Masse
La Protéomique : Etat de l art et perspectives Odile Schiltz Institut de Pharmacologie et de Biologie Structurale CNRS, Université de Toulouse, Odile.Schiltz@ipbs.fr Protéomique et Spectrométrie de Masse
Quantitative proteomics background
 Proteomics data analysis seminar Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post transcriptional regulation of key cellular processes de Groot, M., Daran
Proteomics data analysis seminar Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post transcriptional regulation of key cellular processes de Groot, M., Daran
Appendix 5 Overview of requirements in English
 Appendix 5 Overview of requirements in English This document is a translation of Appendix 4 (Bilag 4) section 2. This translation is meant as a service for the bidder and in case of any differences between
Appendix 5 Overview of requirements in English This document is a translation of Appendix 4 (Bilag 4) section 2. This translation is meant as a service for the bidder and in case of any differences between
Evaluation of LC-MS data for the absolute quantitative analysis of marker proteins
 Evaluation of LC-MS data for the absolute quantitative analysis of marker proteins Nathanaël Delmotte 1, Bettina Mayr 1, Andreas Leinenbach 1, Knut Reinert 2, Oliver Kohlbacher 3, Christoph Klein 4, and
Evaluation of LC-MS data for the absolute quantitative analysis of marker proteins Nathanaël Delmotte 1, Bettina Mayr 1, Andreas Leinenbach 1, Knut Reinert 2, Oliver Kohlbacher 3, Christoph Klein 4, and
Retrospective Analysis of a Host Cell Protein Perfect Storm: Identifying Immunogenic Proteins and Fixing the Problem
 Retrospective Analysis of a Host Cell Protein Perfect Storm: Identifying Immunogenic Proteins and Fixing the Problem Kevin Van Cott, Associate Professor Dept. of Chemical and Biomolecular Engineering Nebraska
Retrospective Analysis of a Host Cell Protein Perfect Storm: Identifying Immunogenic Proteins and Fixing the Problem Kevin Van Cott, Associate Professor Dept. of Chemical and Biomolecular Engineering Nebraska
MS Amanda Standalone User Manual
 MS Amanda Standalone User Manual February 2014 Viktoria Dorfer, Peter Pichler, Stephan Winkler, and Karl Mechtler 1. Installation of MS Amanda Standalone In order to install MS Amanda Standalone please
MS Amanda Standalone User Manual February 2014 Viktoria Dorfer, Peter Pichler, Stephan Winkler, and Karl Mechtler 1. Installation of MS Amanda Standalone In order to install MS Amanda Standalone please
Introduction to mass spectrometry (MS) based proteomics and metabolomics
 based proteomics and metabolomics") Introduction to mass spectrometry (MS) based proteomics and metabolomics Tianwei Yu Department of Biostatistics and Bioinformatics Rollins School of Public Health Emory University September 10, 2015 Background
Introduction to mass spectrometry (MS) based proteomics and metabolomics Tianwei Yu Department of Biostatistics and Bioinformatics Rollins School of Public Health Emory University September 10, 2015 Background
Quantification of Multiple Therapeutic mabs in Serum Using microlc-esi-q-tof Mass Spectrometry
 Quantification of Multiple Therapeutic mabs in Serum Using microlc-esi-q-tof Mass Spectrometry Paula Ladwig, David Barnidge, Mindy Kohlhagen, John Mills, Maria Willrich, Melissa R. Snyder and David Murray
Quantification of Multiple Therapeutic mabs in Serum Using microlc-esi-q-tof Mass Spectrometry Paula Ladwig, David Barnidge, Mindy Kohlhagen, John Mills, Maria Willrich, Melissa R. Snyder and David Murray
--not necessarily a protein! (all proteins are polypeptides, but the converse is not true)
") 00Note Set 5b 1 PEPTIDE BONDS AND POLYPEPTIDES OLIGOPEPTIDE: --chain containing only a few amino acids (see tetrapaptide, Fig 5.9) POLYPEPTIDE CHAINS: --many amino acids joined together --not necessarily
00Note Set 5b 1 PEPTIDE BONDS AND POLYPEPTIDES OLIGOPEPTIDE: --chain containing only a few amino acids (see tetrapaptide, Fig 5.9) POLYPEPTIDE CHAINS: --many amino acids joined together --not necessarily
SimGlycan Software*: A New Predictive Carbohydrate Analysis Tool for MS/MS Data
 SimGlycan Software*: A New Predictive Carbohydrate Analysis Tool for MS/MS Data Automated Data Interpretation for Glycan Characterization Jenny Albanese1, Matthias Glueckmann2 and Christof Lenz2 1Applied
SimGlycan Software*: A New Predictive Carbohydrate Analysis Tool for MS/MS Data Automated Data Interpretation for Glycan Characterization Jenny Albanese1, Matthias Glueckmann2 and Christof Lenz2 1Applied
Mascot Search Results FAQ
 Mascot Search Results FAQ 1 We had a presentation with this same title at our 2005 user meeting. So much has changed in the last 6 years that it seemed like a good idea to re-visit the topic. Just about
Mascot Search Results FAQ 1 We had a presentation with this same title at our 2005 user meeting. So much has changed in the last 6 years that it seemed like a good idea to re-visit the topic. Just about
The Scheduled MRM Algorithm Enables Intelligent Use of Retention Time During Multiple Reaction Monitoring
 The Scheduled MRM Algorithm Enables Intelligent Use of Retention Time During Multiple Reaction Monitoring Delivering up to 2500 MRM Transitions per LC Run Christie Hunter 1, Brigitte Simons 2 1 AB SCIEX,
The Scheduled MRM Algorithm Enables Intelligent Use of Retention Time During Multiple Reaction Monitoring Delivering up to 2500 MRM Transitions per LC Run Christie Hunter 1, Brigitte Simons 2 1 AB SCIEX,
Quantitative mass spectrometry in proteomics: a critical review
 Anal Bioanal Chem (2007) 389:1017 1031 DOI 10.1007/s00216-007-1486-6 REVIEW Quantitative mass spectrometry in proteomics: a critical review Marcus Bantscheff & Markus Schirle & Gavain Sweetman & Jens Rick
Anal Bioanal Chem (2007) 389:1017 1031 DOI 10.1007/s00216-007-1486-6 REVIEW Quantitative mass spectrometry in proteomics: a critical review Marcus Bantscheff & Markus Schirle & Gavain Sweetman & Jens Rick
ALGORITHMS FOR SHOTGUN PROTEOMICS SPECTRAL IDENTIFICATION AND QUALITY ASSESSMENT. Ze-Qiang Ma
 ALGORITHMS FOR SHOTGUN PROTEOMICS SPECTRAL IDENTIFICATION AND QUALITY ASSESSMENT By Ze-Qiang Ma Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment
ALGORITHMS FOR SHOTGUN PROTEOMICS SPECTRAL IDENTIFICATION AND QUALITY ASSESSMENT By Ze-Qiang Ma Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment
In-depth Analysis of Tandem Mass Spectrometry Data from Disparate Instrument Types* S
 Research In-depth Analysis of Tandem Mass Spectrometry Data from Disparate Instrument Types* S Robert J. Chalkley, Peter R. Baker, Katalin F. Medzihradszky, Aenoch J. Lynn, and A. L. Burlingame Mass spectrometric
Research In-depth Analysis of Tandem Mass Spectrometry Data from Disparate Instrument Types* S Robert J. Chalkley, Peter R. Baker, Katalin F. Medzihradszky, Aenoch J. Lynn, and A. L. Burlingame Mass spectrometric
Lecture 13-14 Conformation of proteins Conformation of a protein three-dimensional structure native state. native condition
 Lecture 13-14 Conformation of proteins Conformation of a protein refers to the three-dimensional structure in its native state. There are many different possible conformations for a molecule as large as
Lecture 13-14 Conformation of proteins Conformation of a protein refers to the three-dimensional structure in its native state. There are many different possible conformations for a molecule as large as
A Guide to the Analysis and Purification of Proteins and Peptides by Reversed-Phase HPLC
 A Guide to the Analysis and Purification of Proteins and Peptides by Reversed-Phase HPLC HPLC Columns David Carr Your decision has lasting effects. Choose wisely. HPLC Columns Ultra Inert Base-Deactivated
A Guide to the Analysis and Purification of Proteins and Peptides by Reversed-Phase HPLC HPLC Columns David Carr Your decision has lasting effects. Choose wisely. HPLC Columns Ultra Inert Base-Deactivated
Interpretation of MS-Based Proteomics Data
 Interpretation of MS-Based Proteomics Data Yet-Ran Chen, 陳 逸 然 Agricultural Biotechnology Research Center Academia Sinica Brief Overview of Protein Identification Workflow Protein Sample Specific Protein
Interpretation of MS-Based Proteomics Data Yet-Ran Chen, 陳 逸 然 Agricultural Biotechnology Research Center Academia Sinica Brief Overview of Protein Identification Workflow Protein Sample Specific Protein
Statistics and Algorithms for Peptide Mass Fingerprinting
 Statistics and Algorithms for Peptide Mass Fingerprinting Dipl.-Math. Hans-Michael Kaltenbach Thesis submitted to the Faculty of Technology, Bielefeld University, Germany for the degree of Dr. rer. nat.
Statistics and Algorithms for Peptide Mass Fingerprinting Dipl.-Math. Hans-Michael Kaltenbach Thesis submitted to the Faculty of Technology, Bielefeld University, Germany for the degree of Dr. rer. nat.
itraq Tips and Tricks
 itraq Tips and Tricks Darryl Pappin Cold Spring Harbor Laboratory Patrick Emery Matrix Science Ltd. 1 Good morning. Unfortunately Darryl Pappin is unable to attend the Matrix Science workshop and the ASMS
itraq Tips and Tricks Darryl Pappin Cold Spring Harbor Laboratory Patrick Emery Matrix Science Ltd. 1 Good morning. Unfortunately Darryl Pappin is unable to attend the Matrix Science workshop and the ASMS
Application Note # LCMS-66 Straightforward N-glycopeptide analysis combining fast ion trap data acquisition with new ProteinScape functionalities
 Application Note # LCMS-66 Straightforward N-glycopeptide analysis combining fast ion trap data acquisition with new ProteinScape functionalities Introduction Glycosylation is one of the most common and
Application Note # LCMS-66 Straightforward N-glycopeptide analysis combining fast ion trap data acquisition with new ProteinScape functionalities Introduction Glycosylation is one of the most common and
PeptidomicsDB: a new platform for sharing MS/MS data.
 PeptidomicsDB: a new platform for sharing MS/MS data. Federica Viti, Ivan Merelli, Dario Di Silvestre, Pietro Brunetti, Luciano Milanesi, Pierluigi Mauri NETTAB2010 Napoli, 01/12/2010 Mass Spectrometry
PeptidomicsDB: a new platform for sharing MS/MS data. Federica Viti, Ivan Merelli, Dario Di Silvestre, Pietro Brunetti, Luciano Milanesi, Pierluigi Mauri NETTAB2010 Napoli, 01/12/2010 Mass Spectrometry
Mascot Integra: Data management for Proteomics ASMS 2004
 Mascot Integra: Data management for Proteomics 1 Mascot Integra: Data management for proteomics What is Mascot Integra? What Mascot Integra isn t Instrument integration in Mascot Integra Designing and
Mascot Integra: Data management for Proteomics 1 Mascot Integra: Data management for proteomics What is Mascot Integra? What Mascot Integra isn t Instrument integration in Mascot Integra Designing and
1) Technical informations. - a) How does it work? - b) Purification - c) Quality Control. 2) Standard synthesis
 Technical informations. - a) How does it work? - b) Purification - c) Quality Control. 2) Standard synthesis") 1) Technical informations - a) How does it work? - b) Purification - c) Quality Control 2) Standard synthesis - a) Standard peptides - b) Modified peptides - c) Shipment and Delivery Time - d) How to order?
1) Technical informations - a) How does it work? - b) Purification - c) Quality Control 2) Standard synthesis - a) Standard peptides - b) Modified peptides - c) Shipment and Delivery Time - d) How to order?
STANFORD UNIVERSITY MASS SPECTROMETRY 333 CAMPUS DR., MUDD 175 STANFORD, CA 94305-5080
 Training on the ZQ Open access MS Questions? Contact Dr. Allis Chien allis@stanford.edu 650-723-0710 0710 STANFORD UNIVERSITY MASS SPECTROMETRY STANFORD UNIVERSITY MASS SPECTROMETRY 333 CAMPUS DR., MUDD
Training on the ZQ Open access MS Questions? Contact Dr. Allis Chien allis@stanford.edu 650-723-0710 0710 STANFORD UNIVERSITY MASS SPECTROMETRY STANFORD UNIVERSITY MASS SPECTROMETRY 333 CAMPUS DR., MUDD
Carlos III Networked Proteomics Platform
 Carlos III Networked Proteomics Platform Carlos III Networked Proteomics Platform, ProteoRed-ISCIII is a National Network for the coordination, integration and development of the Spanish Proteomics Facilities
Carlos III Networked Proteomics Platform Carlos III Networked Proteomics Platform, ProteoRed-ISCIII is a National Network for the coordination, integration and development of the Spanish Proteomics Facilities
Investigating Biological Variation of Liver Enzymes in Human Hepatocytes
 Investigating Biological Variation of Liver Enzymes in Human Hepatocytes MS/MS ALL with SWATH Acquisition on the TripleTOF Systems Xu Wang 1, Hui Zhang 2, Christie Hunter 1 1 AB SCIEX, USA, 2 Pfizer, USA
Investigating Biological Variation of Liver Enzymes in Human Hepatocytes MS/MS ALL with SWATH Acquisition on the TripleTOF Systems Xu Wang 1, Hui Zhang 2, Christie Hunter 1 1 AB SCIEX, USA, 2 Pfizer, USA
Data mining with Mascot Integra ASMS 2005
 Data mining with Mascot Integra 1 What is Mascot Integra? Fully functional out-the-box solution for proteomics workflow and data management Support for all the major mass-spectrometry data systems Powered
Data mining with Mascot Integra 1 What is Mascot Integra? Fully functional out-the-box solution for proteomics workflow and data management Support for all the major mass-spectrometry data systems Powered
Metabolomic Profiling of Accurate Mass LC-MS/MS Data to Identify Unexpected Environmental Pollutants
 Metabolomic Profiling of Accurate Mass LC- Data to Identify Unexpected Environmental Pollutants André Schreiber 1, David Cox 1, Nadia Pace 1, Christopher Borton 2 1 AB SCIEX, Concord, ntario, Canada; 2
Metabolomic Profiling of Accurate Mass LC- Data to Identify Unexpected Environmental Pollutants André Schreiber 1, David Cox 1, Nadia Pace 1, Christopher Borton 2 1 AB SCIEX, Concord, ntario, Canada; 2
For the next half hour I m going to be describing some of the different options for peak peaking. The profit is with getting better protein ID or
 For the next half hour I m going to be describing some of the different options for peak peaking. The profit is with getting better protein ID or quantitation, but to be totally honest, the pleasure really
For the next half hour I m going to be describing some of the different options for peak peaking. The profit is with getting better protein ID or quantitation, but to be totally honest, the pleasure really
12/6/12. Dr. Sanjeeva Srivastava. IIT Bombay 2
 Dr. Sanjeeva Srivastava IIT Bombay Case studies 2DE: Drug treatment in malaria parasite Plasma proteome analysis of SARS An overview of DIGE technique Case study DIGE: Serum proteome analysis of prostate
Dr. Sanjeeva Srivastava IIT Bombay Case studies 2DE: Drug treatment in malaria parasite Plasma proteome analysis of SARS An overview of DIGE technique Case study DIGE: Serum proteome analysis of prostate
Application of a New Immobilization H/D Exchange Protocol: A Calmodulin Study
 Application of a New Immobilization H/D Exchange Protocol: A Calmodulin Study Jiang Zhao; Mei Zhu; Daryl E. Gilblin; Michael L. Gross Washington University Center for Biomrdical and Bioorganic Mass Spectrometry:
Application of a New Immobilization H/D Exchange Protocol: A Calmodulin Study Jiang Zhao; Mei Zhu; Daryl E. Gilblin; Michael L. Gross Washington University Center for Biomrdical and Bioorganic Mass Spectrometry:
Q-TOF User s Booklet. Enter your username and password to login. After you login, the data acquisition program will automatically start.
 Q-TOF User s Booklet Enter your username and password to login. After you login, the data acquisition program will automatically start. ~ 1 ~ This data acquisition window will come up after the program
Q-TOF User s Booklet Enter your username and password to login. After you login, the data acquisition program will automatically start. ~ 1 ~ This data acquisition window will come up after the program
Overview. Triple quadrupole (MS/MS) systems provide in comparison to single quadrupole (MS) systems: Introduction
 systems provide in comparison to single quadrupole (MS) systems: Introduction") Advantages of Using Triple Quadrupole over Single Quadrupole Mass Spectrometry to Quantify and Identify the Presence of Pesticides in Water and Soil Samples André Schreiber AB SCIEX Concord, Ontario (Canada)
Advantages of Using Triple Quadrupole over Single Quadrupole Mass Spectrometry to Quantify and Identify the Presence of Pesticides in Water and Soil Samples André Schreiber AB SCIEX Concord, Ontario (Canada)
Database Searching Tutorial/Exercises Jimmy Eng
 Database Searching Tutorial/Exercises Jimmy Eng Use the PETUNIA interface to run a search and generate a pepxml file that is analyzed through the PepXML Viewer. This tutorial will walk you through the
Database Searching Tutorial/Exercises Jimmy Eng Use the PETUNIA interface to run a search and generate a pepxml file that is analyzed through the PepXML Viewer. This tutorial will walk you through the
Separation of Amino Acids by Paper Chromatography
 Separation of Amino Acids by Paper Chromatography Chromatography is a common technique for separating chemical substances. The prefix chroma, which suggests color, comes from the fact that some of the
Separation of Amino Acids by Paper Chromatography Chromatography is a common technique for separating chemical substances. The prefix chroma, which suggests color, comes from the fact that some of the
Pinpointing phosphorylation sites using Selected Reaction Monitoring and Skyline
 Pinpointing phosphorylation sites using Selected Reaction Monitoring and Skyline Christina Ludwig group of Ruedi Aebersold, ETH Zürich The challenge of phosphosite assignment Peptides Phosphopeptides MS/MS
Pinpointing phosphorylation sites using Selected Reaction Monitoring and Skyline Christina Ludwig group of Ruedi Aebersold, ETH Zürich The challenge of phosphosite assignment Peptides Phosphopeptides MS/MS
Molecular Cell Biology. Prof. D. Karunagaran. Department of Biotechnology. Indian Institute of Technology Madras
 Molecular Cell Biology Prof. D. Karunagaran Department of Biotechnology Indian Institute of Technology Madras Module 5 Methods in Cell Biology (Methods to Manipulate Protein, DNA and RNA and Methods to
Molecular Cell Biology Prof. D. Karunagaran Department of Biotechnology Indian Institute of Technology Madras Module 5 Methods in Cell Biology (Methods to Manipulate Protein, DNA and RNA and Methods to
Industry Perspective: Advantages of Open Access and Walkup LC/ MS Supporting Protein Drug Discovery and Development
 Industry Perspective: Advantages of Open Access and Walkup LC/ MS Supporting Protein Drug Discovery and Development Dawn Stickle, Agilent Technologies Originally presented by Eric Fang, Novartis Overview
Industry Perspective: Advantages of Open Access and Walkup LC/ MS Supporting Protein Drug Discovery and Development Dawn Stickle, Agilent Technologies Originally presented by Eric Fang, Novartis Overview
An In-Gel Digestion Protocol
 An In-Gel Digestion Protocol This protocol describes the digestion of a protein present in an SDS-PAGE gel band with trypsin. The band can be taken from either a 1D or 2D electrophoresis gel. Reagents
An In-Gel Digestion Protocol This protocol describes the digestion of a protein present in an SDS-PAGE gel band with trypsin. The band can be taken from either a 1D or 2D electrophoresis gel. Reagents
The Organic Chemistry of Amino Acids, Peptides, and Proteins
 Essential rganic Chemistry Chapter 16 The rganic Chemistry of Amino Acids, Peptides, and Proteins Amino Acids a-amino carboxylic acids. The building blocks from which proteins are made. H 2 N C 2 H Note:
Essential rganic Chemistry Chapter 16 The rganic Chemistry of Amino Acids, Peptides, and Proteins Amino Acids a-amino carboxylic acids. The building blocks from which proteins are made. H 2 N C 2 H Note:
Mass Spectrometry Signal Calibration for Protein Quantitation
 Cambridge Isotope Laboratories, Inc. www.isotope.com Proteomics Mass Spectrometry Signal Calibration for Protein Quantitation Michael J. MacCoss, PhD Associate Professor of Genome Sciences University of
Cambridge Isotope Laboratories, Inc. www.isotope.com Proteomics Mass Spectrometry Signal Calibration for Protein Quantitation Michael J. MacCoss, PhD Associate Professor of Genome Sciences University of