Proteomics software available in the public domain. Pratik Jagtap Minnesota Supercomputing institute
|
|
|
- Cecil Marshall
- 7 years ago
- Views:
Transcription
1 Proteomics software available in the public domain. Pratik Jagtap Minnesota Supercomputing institute

2 Two-Dimensional gel electrophoresis pi Mw Proteins are resolved based on their isolelectric point (using isoelectric focusing) and then molecular weight (using SDS-PAGE). Gels are compared, differentially expressed proteins are excised and identified.

3 Proteomics Fifteen Years Ago
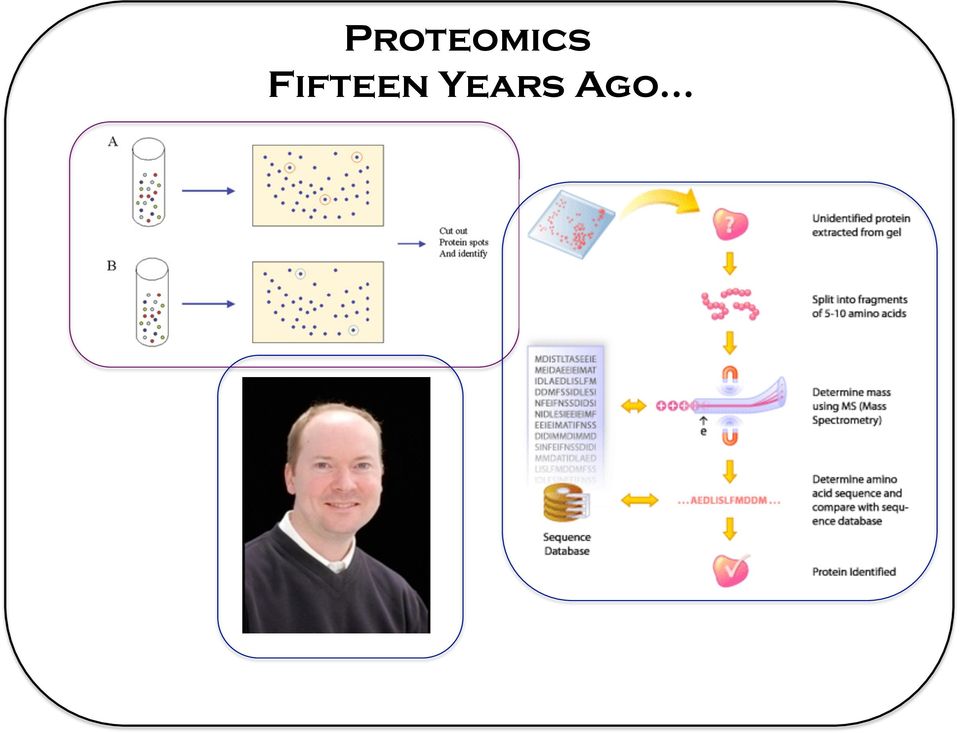
4 Proteomics Fifteen Years Ago Mass Spectrometry Data Extrac5on. Search algorithm Analysis So9ware that correlates the protein ID to the excised gel spot.

5 Two-Dimensional gel electrophoresis pi Mw 2DGE : High molecular weight proteins, low molecular weight proteins, proteins with extreme isoelectric points, membrane proteins were underrepresented in the analysis.

6 Multi-Dimensional Protein Identification Technology

7 Proteomics workflow Protein Peptide Fragmentation Mass spectrum Search against database.

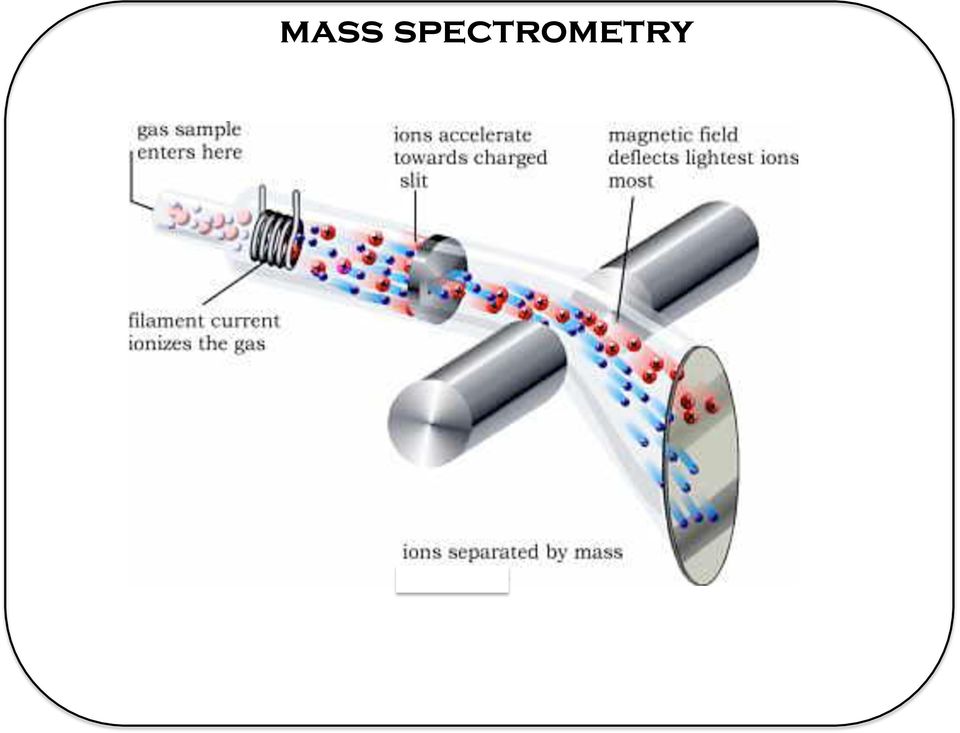
8 mass spectrometry
9 Mass Spectrometers & data formats Thermofinnigan Xcalibur /.raw Life Technologies Analyst /.wiff ;.t2d Sequest.dta.out ProteinPilot.t2d.group Waters Masslynx /.raw Bruker.baf mzxml pepxml mzml mzdata protxml X! tandem.xml OMSSA.xml.omx Mascot.mgf.dat

10 Proteo-Informatics Mass Spectrometry Data Extrac5on. Data Conversion. Search algorithm De novo Tools. Sta5s5cal valida5on of pep5de and protein iden5fica5ons. Quan5ta5ve Tools. Targeted Proteomics Spectral Matching Data Dissemina5on

11 Mass Spectrometry Data Extrac5on. Data extraction ReAdW ReAdW converts Xcalibur.raw files to universal mzxml format. T2D Extractor A tool that can access the Applied Biosystem s MALDI-TOF/TOF 4700 and 4800 database and can extract T2D files as well as peak lists. It can be used to extract individual spectra, runs, or entire spotsets. MS/MS peaklists are provided in.mgf formats. Runs on Java 1.5 platform. LCMS Peaklist Extractor Batch mode tool for extracting concatenated.mgf peaklist files. Quantitation Extractor Batch mode tool for extracting areas for peaks in MS/MS spectra.

12 Mass Spectrometers & data formats Thermofinnigan Xcalibur /.raw Life Technologies Analyst /.wiff ;.t2d Waters Masslynx /.raw Bruker.baf mzxml pepxml mzml mzdata protxml Mass Spectrometry Mascot.mgf.dat Sequest.dta.out X! tandem.xml OMSSA.xml.omx Data Conversion.

13 Mass Spectrometry Data Conversion. data conversion mzxml2other Converter from mzxml to sequest dta, mascot generic and micromass pkl formats. Peak List Conversion Utility (Java Web Start) The ProteomeCommons.org IO Framework's tool for converting peak list and spectrum files between different formats. The tool can also merge multiple peak lists into a single concatinated peak list. The tools uses Java Web Start and runs locally on your computer. /MM_File_Conversion_1p0.exe MassMatrix File Conversion Tools These tools convert between common input formats:.raw,.mzxml,.mgf.

14 search algorithm Mass Spectrometry Data Extrac5on. Data Conversion. Search algorithm

15 Search algorithm SEARCH ALGORITHM

16 Search algorithm X!tandem & the GPM X! Tandem can be utilized as a web-based application or deployed locally using precompiled binaries and FASTA-formatted files. X!Tandem takes inputs in.xml format and outputs.xml format. The data analysis components consist of Input file ; FASTA, Taxonomy; Parameters and output. Central Axiom : For each identifiable protein, there is at least one detectable tryptic peptide. Extensively search for modified/ non-enzymatic peptides only on identified proteins. How far is the top-scoring match from the rest of the pack? Uses E-value. Much faster than Sequest s Xcorr. The Global Proteome Machine Organization X!Hunter X! P3 Common

17 Search algorithm OMSSA OMSSA takes experimental ms/ms spectra, filters noise peaks, extracts m/z values, and then compares these m/z values to calculated m/z values derived from peptides produced by an in silico digestion of a protein sequence library. Calculates E-value as a discriminant score. An E-value for a hit is a score that is the expected number of random hits from a search library to a given spectrum such that the random hits have an equal or better score than the hit. It uses classical hypothesis testing based on type of statistical model that is used in BLAST. Faster; Runs on all platforms

18 Search algorithm maxquant MaxQuant is an integrated suite of algorithms specifically developed for highresolution, quantitative MS data. MaxQuant detects peaks, isotope clusters and stable amino acid isotope-labeled (SILAC) peptide pairs as three-dimensional objects in m/z, elution time and signal intensity space. By integrating multiple mass measurements, mass accuracy in the p.p.b. range is achieved. MaxQuant quantifies several hundred thousand peptides per SILAC-proteome experiment.
 peptide pairs as three-dimensional")
19 De novo tools Mass Spectrometry Data Extrac5on. Data Conversion. Search algorithm De novo Tools.

20 De novo Tools. de novo analysis Protein Peptide Fragmentation Mass spectrum Search against database. De novo Analysis : Generate sequence from spectrum and match against database by using BLAST

21 De novo Tools. pepnovo hep://pep5de.ucsd.edu/pepnovo.html PepNovo is a software for de novo sequencing of peptides from mass spectra. PepNovo uses a probabilistic network to model the peptide fragmentation events in a mass spectrometer. In addition, it uses a likelihood ratio hypothesis test to determine if the peaks observed in the mass spectrum are more likely to have been produced under the fragmentation model, than under a probabilistic model that treats the appearance of peaks as random events.
22 De novo Tools. lutefisk LUTEFISK uses a graph theory approach for de novo peptide sequence determinations from low-energy collision-induced dissociation (CID) data of tryptic peptides. Lutefisk converts all of the ions into their corresponding b-ion masses by making N- and C-terminal evidence lists that contain evidence for cleavage at every possible b-ion mass. Once the sequence spectrum has been established, the program proceeds by tracing sequences starting at the N-terminus. Highest ranked sequences are subjected to a cross-correlation analysis and scores are combined and normalized to produce a final score and ranking.
23 spectral matching Mass Spectrometry Data Extrac5on. Data Conversion. Search algorithm De novo Tools. Spectral Matching
24 Spectral Matching x!hunter X! Hunter is a search engine that compares experimentally observed spectra directly with a library of spectra that have been confidently assigned to a particular peptide sequence (an Annotated Spectrum Library, or ASL). It can identify proteins using information from large number of spectra in GPMDB database. Creation of ASLs : 1) Confident assignments for human and yeast peptides were extracted from GPMDB. 2) Replicate observations of the same peptide were averaged together and a final list of averaged peptide spectra was produced. Because the sequence modifications and cleavage sites for the peptides in the sequence library are already known, it is not necessary to specify as many parameters for this type of search as in more conventional search engines. This type of pattern matching tool is ideal for applications such as biomarker discovery.
25 Spectral Matching MS-Clustering MS-Clustering of MS/MS spectra takes advantage of dataset redundancy by identifying multiple spectra of the same peptide and replacing them with a single representative spectrum. Analyzing only representative spectra results in significant speed-up of MS/MS database searches. Large MS/MS data sets (over 10 million spectra) were reduced to smaller datasets and resulted in higher number of peptide identifications as compared to regular nonclustered searches.
26 Mass Spectrometry Data Extrac5on. Data Conversion. Search algorithm De novo Tools. Spectral Matching Sta5s5cal valida5on of pep5de and protein iden5fica5ons.
27 Sta5s5cal valida5on of pep5de and protein iden5fica5ons.
28 Trans-proteomic pipeline Sta5s5cal valida5on of pep5de and protein iden5fica5ons. Trans-Proteomic Pipeline (TPP) is a data analysis pipeline for the analysis of LC/ MS/MS proteomics data. TPP includes modules for validation of database search results, quantitation of isotopically labeled samples, and validation of protein identifications, as well as tools for viewing raw LC/MS data, peptide identification results, and protein identification results. The XML backbone of this pipeline enables a uniform analysis for LC/MS/MS data generated by a wide variety of mass spectrometer types, and assigned peptides using a wide variety of database search engines.
29 peparml Sta5s5cal valida5on of pep5de and protein iden5fica5ons. X!Tandem Mascot Feature extraction PepArML OMSSA Other A model-free, result-combining peptide identification arbiter via machine learning.
30 Quantitative tools Mass Spectrometry Data Extrac5on. Data Conversion. Search algorithm De novo Tools. Sta5s5cal valida5on of pep5de and protein iden5fica5ons. Quan5ta5ve Tools. Spectral Matching
31 itraq : Isobaric Tags for Relative and Absolute Quantification. Isobaric Tag (Total mass = 145) Reporter Charged Balance Neutral loss Peptide Reactive Group Trypsin digest PRG PRG PRG PRG Mix MS -N H -N H -N H -N H MS [Reporter-Balance-Peptide] MS/MS % Intensity % Intensity Mass (m/z) QGQPIGLGEASNDTWI TTK Mass (m/z)
32 Proteomics Quantitatition
33 Quan5ta5ve Tools. i-tracker i-tracker is an open-source peptide quantitation algorithm that allows the user to extract reporter ion peak ratios from non-centroided peak lists. The algorithm uses.dta and.mgf files as inputs. The reporter ion areas are calculated and corrected for their purity. The.csv output of i-tracker allows for the relative comparison of the itraq labeled peptides.
34 Quan5ta5ve Tools. TPP Quantitative Tools ASAP ratio, Xpress and libra ASAPRatio Automated Statistical Analysis on Protein Ratio (ASAPRatio) accurately calculates the relative abundances of proteins and the corresponding confidence intervals from ICATtype ESI-LC/MS data. XPRESS The XPRESS software calculates the relative abundance of proteins, such as those obtained from an ICAT-reagent labeled experiment, by reconstructing the light and heavy elution profiles of the precursor ions and determining the elution areas of each peak. LIBRA Libra is a module within the trans-proteomic pipeline to perform quantification on MS/MS spectra that have itraq labeled samples.
35 APEX Quan5ta5ve Tools. The APEX Quantitative Proteomics Tool is a free and open source Java implementation of the APEX technique for the absolute quantitation of proteins based on standard LC- MS/MS proteomics data. It uses machine learning techniques to improve quantitation accuracy for labelfree technique. The APEX Tool provides an intuitive user interface, an integrated help system, and rich documentation. A tutorial and sample data set is included to help first time users become acquainted with the system.
36 Quan5ta5ve Tool maxquant MaxQuant quantifies several hundred thousand peptides per SILAC-proteome experiment.
37 Targeted Proteomics Mass Spectrometry Data Extrac5on. Data Conversion. Search algorithm De novo Tools. Sta5s5cal valida5on of pep5de and protein iden5fica5ons. Quan5ta5ve Tools. Targeted Proteomics Spectral Matching
38 Targeted Proteomics Biochemistry vs Proteomics Targeted proteomics vs Shotgun Proteomics
39 Targeted Proteomics MRM Selectivity, Sensitivity and Dynamic Range Quantitative Proteomics Results Prediction Choose and Optimize Transistions
40 Targeted Proteomics TIQAM TIQAM generates MRM transition lists and identifies the best performing transitions from MRM pre-experiments. In addition TIQAM provides a viewer to validate transitions by MRM-triggered MS/MS experiments. All the peptide and transition information is stored in a database to enable smart retrieval of the validated transitions for quantitative analysis. Commercial softwares : MRMPilot (Applied Biosystems), SRM Workflow Software (Thermo Scientific), VerifyE (Waters) and Optimizer (Agilent Technologies).
41 Targeted Proteomics X! P3 ftp://ftp.thegpm.org/proteotypic_peptide_profiles Uses identification of proteotypic peptides for identification of a protein. Because there will only be a few proteotypic peptides for a protein, it improves both the speed and accuracy of the resultant protein identifications. The X! P3 (Proteotypic Peptide Profiler) project uses the following steps : 1. In the first round, the spectrum data set is examined for the presence of proteotypic peptides. This is done by querying GPMDB to find the best peptides representative of a particular protein. 2. The full protein sequences of the proteins identified in the first round are then pulled from a sequence library. 3. Using this small set of full sequences, multiple rounds of refinement are performed to extract all of the non-proteotypic peptides from the full spectrum data set An X! P3 server has been established for two model organisms, namely Homo sapiens and Saccharomyces cerevisiae, as well as several commonly observed experimental artifacts, such as BSA and trypsin.
42 Mass Spectrometry Data Extrac5on. Data Conversion. Search algorithm De novo Tools. Spectral Matching Sta5s5cal valida5on of pep5de and protein iden5fica5ons. Quan5ta5ve Tools. Data Dissemina5on Targeted Proteomics Your Answer is going to be determined by the ques5on asked.
43 Data Dissemina5on Prestomic An open-source suite of tools for storing data and for presenting the data in a user-friendly format via a browser. The program was developed using mostly Perl.
44 Data Dissemina5on Tranche Tranche is a free and open source file sharing tool that enables the storage of large amounts of data. Designed and built with scientists and researchers in mind, Tranche can handle very large data sets, is secure, is scalable, and all data sets are citable in scientific journals.
45 Proteomic pipelines that use Open-source software. CPAS Open source toolkit that integrates open source proteomics tools along with existing commercial software. CORRA Statistical Analysis tools for Quantitative proteomics SysPIMP Identify mutated proteins from mass spectrometry results. SwissPIT Multitool platform that promotes use of multiple search algorithms. mmass data miner The OpenMS Proteomics Pipeline
46 Protip Raw Data from Orbitrap mzxml format Mgf format dta format X!TANDEM search OMSSA search SEQUEST search MASCOT search Scaffold Analysis Scaffold Viewer
47
48 performing multiple searches through Protip HUMAN DATASET mzxml format Mgf format # of peptides X!TANDEM search OMSSA search 491 dta format SEQUEST search MASCOT search 462 # of proteins Sequest X! tandem Mascot All Together Scaffold Analysis Sequest + Mascot Sequest + X! tandem X! tandem + Mascot
49 LAST WORD Questions? Pratik Jagtap
Proteomic data analysis for Orbitrap datasets using Resources available at MSI. September 28 th 2011 Pratik Jagtap
 Proteomic data analysis for Orbitrap datasets using Resources available at MSI. September 28 th 2011 Pratik Jagtap The Minnesota http://www.mass.msi.umn.edu/ Proteomics workflow Trypsin Protein Peptides
Proteomic data analysis for Orbitrap datasets using Resources available at MSI. September 28 th 2011 Pratik Jagtap The Minnesota http://www.mass.msi.umn.edu/ Proteomics workflow Trypsin Protein Peptides
泛 用 蛋 白 質 體 學 之 質 譜 儀 資 料 分 析 平 台 的 建 立 與 應 用 Universal Mass Spectrometry Data Analysis Platform for Quantitative and Qualitative Proteomics
 泛 用 蛋 白 質 體 學 之 質 譜 儀 資 料 分 析 平 台 的 建 立 與 應 用 Universal Mass Spectrometry Data Analysis Platform for Quantitative and Qualitative Proteomics 2014 Training Course Wei-Hung Chang ( 張 瑋 宏 ) ABRC, Academia
泛 用 蛋 白 質 體 學 之 質 譜 儀 資 料 分 析 平 台 的 建 立 與 應 用 Universal Mass Spectrometry Data Analysis Platform for Quantitative and Qualitative Proteomics 2014 Training Course Wei-Hung Chang ( 張 瑋 宏 ) ABRC, Academia
Tutorial for Proteomics Data Submission. Katalin F. Medzihradszky Robert J. Chalkley UCSF
 Tutorial for Proteomics Data Submission Katalin F. Medzihradszky Robert J. Chalkley UCSF Why Have Guidelines? Large-scale proteomics studies create huge amounts of data. It is impossible/impractical to
Tutorial for Proteomics Data Submission Katalin F. Medzihradszky Robert J. Chalkley UCSF Why Have Guidelines? Large-scale proteomics studies create huge amounts of data. It is impossible/impractical to
ProteinScape. Innovation with Integrity. Proteomics Data Analysis & Management. Mass Spectrometry
 ProteinScape Proteomics Data Analysis & Management Innovation with Integrity Mass Spectrometry ProteinScape a Virtual Environment for Successful Proteomics To overcome the growing complexity of proteomics
ProteinScape Proteomics Data Analysis & Management Innovation with Integrity Mass Spectrometry ProteinScape a Virtual Environment for Successful Proteomics To overcome the growing complexity of proteomics
ProteinPilot Report for ProteinPilot Software
 ProteinPilot Report for ProteinPilot Software Detailed Analysis of Protein Identification / Quantitation Results Automatically Sean L Seymour, Christie Hunter SCIEX, USA Pow erful mass spectrometers like
ProteinPilot Report for ProteinPilot Software Detailed Analysis of Protein Identification / Quantitation Results Automatically Sean L Seymour, Christie Hunter SCIEX, USA Pow erful mass spectrometers like
using ms based proteomics
 quantification using ms based proteomics lennart martens Computational Omics and Systems Biology Group Department of Medical Protein Research, VIB Department of Biochemistry, Ghent University Ghent, Belgium
quantification using ms based proteomics lennart martens Computational Omics and Systems Biology Group Department of Medical Protein Research, VIB Department of Biochemistry, Ghent University Ghent, Belgium
Mass Spectrometry Based Proteomics
 Mass Spectrometry Based Proteomics Proteomics Shared Research Oregon Health & Science University Portland, Oregon This document is designed to give a brief overview of Mass Spectrometry Based Proteomics
Mass Spectrometry Based Proteomics Proteomics Shared Research Oregon Health & Science University Portland, Oregon This document is designed to give a brief overview of Mass Spectrometry Based Proteomics
Aiping Lu. Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn
 Aiping Lu Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn Proteome and Proteomics PROTEin complement expressed by genome Marc Wilkins Electrophoresis. 1995. 16(7):1090-4. proteomics
Aiping Lu Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn Proteome and Proteomics PROTEin complement expressed by genome Marc Wilkins Electrophoresis. 1995. 16(7):1090-4. proteomics
MRMPilot Software: Accelerating MRM Assay Development for Targeted Quantitative Proteomics
 MRMPilot Software: Accelerating MRM Assay Development for Targeted Quantitative Proteomics With Unique QTRAP and TripleTOF 5600 System Technology Targeted peptide quantification is a rapidly growing application
MRMPilot Software: Accelerating MRM Assay Development for Targeted Quantitative Proteomics With Unique QTRAP and TripleTOF 5600 System Technology Targeted peptide quantification is a rapidly growing application
Global and Discovery Proteomics Lecture Agenda
 Global and Discovery Proteomics Christine A. Jelinek, Ph.D. Johns Hopkins University School of Medicine Department of Pharmacology and Molecular Sciences Middle Atlantic Mass Spectrometry Laboratory Global
Global and Discovery Proteomics Christine A. Jelinek, Ph.D. Johns Hopkins University School of Medicine Department of Pharmacology and Molecular Sciences Middle Atlantic Mass Spectrometry Laboratory Global
Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench
 Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench Application Guide Agilent Technologies Notices Agilent Technologies, Inc. 2012 No part of this manual may be reproduced in any form or by any
Agilent G2721AA/G2733AA Spectrum Mill MS Proteomics Workbench Application Guide Agilent Technologies Notices Agilent Technologies, Inc. 2012 No part of this manual may be reproduced in any form or by any
Database Searching Tutorial/Exercises Jimmy Eng
 Database Searching Tutorial/Exercises Jimmy Eng Use the PETUNIA interface to run a search and generate a pepxml file that is analyzed through the PepXML Viewer. This tutorial will walk you through the
Database Searching Tutorial/Exercises Jimmy Eng Use the PETUNIA interface to run a search and generate a pepxml file that is analyzed through the PepXML Viewer. This tutorial will walk you through the
Master course KEMM03 Principles of Mass Spectrometric Protein Characterization. Exam
 Exam Master course KEMM03 Principles of Mass Spectrometric Protein Characterization 2010-10-29 kl 08.15-13.00 Use a new paper for answering each question! Write your name on each paper! Aids: Mini calculator,
Exam Master course KEMM03 Principles of Mass Spectrometric Protein Characterization 2010-10-29 kl 08.15-13.00 Use a new paper for answering each question! Write your name on each paper! Aids: Mini calculator,
PeptidomicsDB: a new platform for sharing MS/MS data.
 PeptidomicsDB: a new platform for sharing MS/MS data. Federica Viti, Ivan Merelli, Dario Di Silvestre, Pietro Brunetti, Luciano Milanesi, Pierluigi Mauri NETTAB2010 Napoli, 01/12/2010 Mass Spectrometry
PeptidomicsDB: a new platform for sharing MS/MS data. Federica Viti, Ivan Merelli, Dario Di Silvestre, Pietro Brunetti, Luciano Milanesi, Pierluigi Mauri NETTAB2010 Napoli, 01/12/2010 Mass Spectrometry
Challenges in Computational Analysis of Mass Spectrometry Data for Proteomics
 Ma B. Challenges in computational analysis of mass spectrometry data for proteomics. SCIENCE AND TECHNOLOGY 25(1): 1 Jan. 2010 JOURNAL OF COMPUTER Challenges in Computational Analysis of Mass Spectrometry
Ma B. Challenges in computational analysis of mass spectrometry data for proteomics. SCIENCE AND TECHNOLOGY 25(1): 1 Jan. 2010 JOURNAL OF COMPUTER Challenges in Computational Analysis of Mass Spectrometry
Introduction to Proteomics
 Introduction to Proteomics Åsa Wheelock, Ph.D. Division of Respiratory Medicine & Karolinska Biomics Center asa.wheelock@ki.se In: Systems Biology and the Omics Cascade, Karolinska Institutet, June 9-13,
Introduction to Proteomics Åsa Wheelock, Ph.D. Division of Respiratory Medicine & Karolinska Biomics Center asa.wheelock@ki.se In: Systems Biology and the Omics Cascade, Karolinska Institutet, June 9-13,
MultiQuant Software 2.0 for Targeted Protein / Peptide Quantification
 MultiQuant Software 2.0 for Targeted Protein / Peptide Quantification Gold Standard for Quantitative Data Processing Because of the sensitivity, selectivity, speed and throughput at which MRM assays can
MultiQuant Software 2.0 for Targeted Protein / Peptide Quantification Gold Standard for Quantitative Data Processing Because of the sensitivity, selectivity, speed and throughput at which MRM assays can
Already said. Already said. Outlook. Look at LC-MS data. A look at data for quantitative analysis using MSight and Phenyx. What data for quantitation?
 A look at data for quantitative analysis using MSight and Phenyx Pierre-Alain Binz Institut Suisse de Bioinformatique GeneBio SA Atelier Protéomique Quantitative 25-27 Juin 2007 La Grande Motte Already
A look at data for quantitative analysis using MSight and Phenyx Pierre-Alain Binz Institut Suisse de Bioinformatique GeneBio SA Atelier Protéomique Quantitative 25-27 Juin 2007 La Grande Motte Already
Session 1. Course Presentation: Mass spectrometry-based proteomics for molecular and cellular biologists
 Program Overview Session 1. Course Presentation: Mass spectrometry-based proteomics for molecular and cellular biologists Session 2. Principles of Mass Spectrometry Session 3. Mass spectrometry based proteomics
Program Overview Session 1. Course Presentation: Mass spectrometry-based proteomics for molecular and cellular biologists Session 2. Principles of Mass Spectrometry Session 3. Mass spectrometry based proteomics
CPAS Overview. Josh Eckels LabKey Software jeckels@labkey.com
 CPAS Overview Josh Eckels LabKey Software jeckels@labkey.com CPAS Web-based system for processing, storing, and analyzing results of MS/MS experiments Key goals: Provide a great analysis front-end for
CPAS Overview Josh Eckels LabKey Software jeckels@labkey.com CPAS Web-based system for processing, storing, and analyzing results of MS/MS experiments Key goals: Provide a great analysis front-end for
The Scheduled MRM Algorithm Enables Intelligent Use of Retention Time During Multiple Reaction Monitoring
 The Scheduled MRM Algorithm Enables Intelligent Use of Retention Time During Multiple Reaction Monitoring Delivering up to 2500 MRM Transitions per LC Run Christie Hunter 1, Brigitte Simons 2 1 AB SCIEX,
The Scheduled MRM Algorithm Enables Intelligent Use of Retention Time During Multiple Reaction Monitoring Delivering up to 2500 MRM Transitions per LC Run Christie Hunter 1, Brigitte Simons 2 1 AB SCIEX,
Sub menu of functions to give the user overall information about the data in the file
 Visualize The Multitool for Proteomics! File Open Opens an.ez2 file to be examined. Import from TPP Imports data from files created by Trans Proteomic Pipeline. User chooses mzxml, pepxml and FASTA files
Visualize The Multitool for Proteomics! File Open Opens an.ez2 file to be examined. Import from TPP Imports data from files created by Trans Proteomic Pipeline. User chooses mzxml, pepxml and FASTA files
MASCOT Search Results Interpretation
 The Mascot protein identification program (Matrix Science, Ltd.) uses statistical methods to assess the validity of a match. MS/MS data is not ideal. That is, there are unassignable peaks (noise) and usually
The Mascot protein identification program (Matrix Science, Ltd.) uses statistical methods to assess the validity of a match. MS/MS data is not ideal. That is, there are unassignable peaks (noise) and usually
For the next half hour I m going to be describing some of the different options for peak peaking. The profit is with getting better protein ID or
 For the next half hour I m going to be describing some of the different options for peak peaking. The profit is with getting better protein ID or quantitation, but to be totally honest, the pleasure really
For the next half hour I m going to be describing some of the different options for peak peaking. The profit is with getting better protein ID or quantitation, but to be totally honest, the pleasure really
OpenMS A Framework for Quantitative HPLC/MS-Based Proteomics
 OpenMS A Framework for Quantitative HPLC/MS-Based Proteomics Knut Reinert 1, Oliver Kohlbacher 2,Clemens Gröpl 1, Eva Lange 1, Ole Schulz-Trieglaff 1,Marc Sturm 2 and Nico Pfeifer 2 1 Algorithmische Bioinformatik,
OpenMS A Framework for Quantitative HPLC/MS-Based Proteomics Knut Reinert 1, Oliver Kohlbacher 2,Clemens Gröpl 1, Eva Lange 1, Ole Schulz-Trieglaff 1,Marc Sturm 2 and Nico Pfeifer 2 1 Algorithmische Bioinformatik,
ProSightPC 3.0 Quick Start Guide
 ProSightPC 3.0 Quick Start Guide The Thermo ProSightPC 3.0 application is the only proteomics software suite that effectively supports high-mass-accuracy MS/MS experiments performed on LTQ FT and LTQ Orbitrap
ProSightPC 3.0 Quick Start Guide The Thermo ProSightPC 3.0 application is the only proteomics software suite that effectively supports high-mass-accuracy MS/MS experiments performed on LTQ FT and LTQ Orbitrap
Introduction to Proteomics 1.0
 Introduction to Proteomics 1.0 CMSP Workshop Tim Griffin Associate Professor, BMBB Faculty Director, CMSP Objectives Why are we here? For participants: Learn basics of MS-based proteomics Learn what s
Introduction to Proteomics 1.0 CMSP Workshop Tim Griffin Associate Professor, BMBB Faculty Director, CMSP Objectives Why are we here? For participants: Learn basics of MS-based proteomics Learn what s
Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance?
 Optimization 1 Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance? Where to begin? 2 Sequence Databases Swiss-prot MSDB, NCBI nr dbest Species specific ORFS
Optimization 1 Choices, choices, choices... Which sequence database? Which modifications? What mass tolerance? Where to begin? 2 Sequence Databases Swiss-prot MSDB, NCBI nr dbest Species specific ORFS
Introduction to Database Searching using MASCOT
 Introduction to Database Searching using MASCOT 1 Three ways to use mass spectrometry data for protein identification 1.Peptide Mass Fingerprint A set of peptide molecular masses from an enzyme digest
Introduction to Database Searching using MASCOT 1 Three ways to use mass spectrometry data for protein identification 1.Peptide Mass Fingerprint A set of peptide molecular masses from an enzyme digest
In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates
 In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates Using the Explore Workflow on the AB SCIEX TripleTOF 5600 System A major challenge in proteomics
In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates Using the Explore Workflow on the AB SCIEX TripleTOF 5600 System A major challenge in proteomics
Protein Prospector and Ways of Calculating Expectation Values
 Protein Prospector and Ways of Calculating Expectation Values 1/16 Aenoch J. Lynn; Robert J. Chalkley; Peter R. Baker; Mark R. Segal; and Alma L. Burlingame University of California, San Francisco, San
Protein Prospector and Ways of Calculating Expectation Values 1/16 Aenoch J. Lynn; Robert J. Chalkley; Peter R. Baker; Mark R. Segal; and Alma L. Burlingame University of California, San Francisco, San
Proteomic Analysis using Accurate Mass Tags. Gordon Anderson PNNL January 4-5, 2005
 Proteomic Analysis using Accurate Mass Tags Gordon Anderson PNNL January 4-5, 2005 Outline Accurate Mass and Time Tag (AMT) based proteomics Instrumentation Data analysis Data management Challenges 2 Approach
Proteomic Analysis using Accurate Mass Tags Gordon Anderson PNNL January 4-5, 2005 Outline Accurate Mass and Time Tag (AMT) based proteomics Instrumentation Data analysis Data management Challenges 2 Approach
Tutorial 9: SWATH data analysis in Skyline
 Tutorial 9: SWATH data analysis in Skyline In this tutorial we will learn how to perform targeted post-acquisition analysis for protein identification and quantitation using a data-independent dataset
Tutorial 9: SWATH data analysis in Skyline In this tutorial we will learn how to perform targeted post-acquisition analysis for protein identification and quantitation using a data-independent dataset
Quantitative proteomics background
 Proteomics data analysis seminar Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post transcriptional regulation of key cellular processes de Groot, M., Daran
Proteomics data analysis seminar Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post transcriptional regulation of key cellular processes de Groot, M., Daran
Effects of Intelligent Data Acquisition and Fast Laser Speed on Analysis of Complex Protein Digests
 Effects of Intelligent Data Acquisition and Fast Laser Speed on Analysis of Complex Protein Digests AB SCIEX TOF/TOF 5800 System with DynamicExit Algorithm and ProteinPilot Software for Robust Protein
Effects of Intelligent Data Acquisition and Fast Laser Speed on Analysis of Complex Protein Digests AB SCIEX TOF/TOF 5800 System with DynamicExit Algorithm and ProteinPilot Software for Robust Protein
AB SCIEX TOF/TOF 4800 PLUS SYSTEM. Cost effective flexibility for your core needs
 AB SCIEX TOF/TOF 4800 PLUS SYSTEM Cost effective flexibility for your core needs AB SCIEX TOF/TOF 4800 PLUS SYSTEM It s just what you expect from the industry leader. The AB SCIEX 4800 Plus MALDI TOF/TOF
AB SCIEX TOF/TOF 4800 PLUS SYSTEM Cost effective flexibility for your core needs AB SCIEX TOF/TOF 4800 PLUS SYSTEM It s just what you expect from the industry leader. The AB SCIEX 4800 Plus MALDI TOF/TOF
Error Tolerant Searching of Uninterpreted MS/MS Data
 Error Tolerant Searching of Uninterpreted MS/MS Data 1 In any search of a large LC-MS/MS dataset 2 There are always a number of spectra which get poor scores, or even no match at all. 3 Sometimes, this
Error Tolerant Searching of Uninterpreted MS/MS Data 1 In any search of a large LC-MS/MS dataset 2 There are always a number of spectra which get poor scores, or even no match at all. 3 Sometimes, this
Application Note # LCMS-81 Introducing New Proteomics Acquisiton Strategies with the compact Towards the Universal Proteomics Acquisition Method
 Application Note # LCMS-81 Introducing New Proteomics Acquisiton Strategies with the compact Towards the Universal Proteomics Acquisition Method Introduction During the last decade, the complexity of samples
Application Note # LCMS-81 Introducing New Proteomics Acquisiton Strategies with the compact Towards the Universal Proteomics Acquisition Method Introduction During the last decade, the complexity of samples
Increasing the Multiplexing of High Resolution Targeted Peptide Quantification Assays
 Increasing the Multiplexing of High Resolution Targeted Peptide Quantification Assays Scheduled MRM HR Workflow on the TripleTOF Systems Jenny Albanese, Christie Hunter AB SCIEX, USA Targeted quantitative
Increasing the Multiplexing of High Resolution Targeted Peptide Quantification Assays Scheduled MRM HR Workflow on the TripleTOF Systems Jenny Albanese, Christie Hunter AB SCIEX, USA Targeted quantitative
Proteomics in Practice
 Reiner Westermeier, Torn Naven Hans-Rudolf Höpker Proteomics in Practice A Guide to Successful Experimental Design 2008 Wiley-VCH Verlag- Weinheim 978-3-527-31941-1 Preface Foreword XI XIII Abbreviations,
Reiner Westermeier, Torn Naven Hans-Rudolf Höpker Proteomics in Practice A Guide to Successful Experimental Design 2008 Wiley-VCH Verlag- Weinheim 978-3-527-31941-1 Preface Foreword XI XIII Abbreviations,
Research-grade Targeted Proteomics Assay Development: PRMs for PTM Studies with Skyline or, How I learned to ditch the triple quad and love the QE
 Research-grade Targeted Proteomics Assay Development: PRMs for PTM Studies with Skyline or, How I learned to ditch the triple quad and love the QE Jacob D. Jaffe Skyline Webinar July 2015 Proteomics and
Research-grade Targeted Proteomics Assay Development: PRMs for PTM Studies with Skyline or, How I learned to ditch the triple quad and love the QE Jacob D. Jaffe Skyline Webinar July 2015 Proteomics and
Absolute quantification of low abundance proteins by shotgun proteomics
 Absolute quantification of low abundance proteins by shotgun proteomics Dr. Stefanie Wienkoop www.proteomefactory.com In cooperation with: Max-Planck-Institut für Molekulare Pflanzenphysiologie Stable
Absolute quantification of low abundance proteins by shotgun proteomics Dr. Stefanie Wienkoop www.proteomefactory.com In cooperation with: Max-Planck-Institut für Molekulare Pflanzenphysiologie Stable
Mascot Integra: Data management for Proteomics ASMS 2004
 Mascot Integra: Data management for Proteomics 1 Mascot Integra: Data management for proteomics What is Mascot Integra? What Mascot Integra isn t Instrument integration in Mascot Integra Designing and
Mascot Integra: Data management for Proteomics 1 Mascot Integra: Data management for proteomics What is Mascot Integra? What Mascot Integra isn t Instrument integration in Mascot Integra Designing and
How Mascot Integra helps run a Core Lab
 How Mascot Integra helps run a Core Lab 1 Areas where a database can help a core lab Project, experiment and sample tracking Flexibility in experiment design Role based security Automation Custom results
How Mascot Integra helps run a Core Lab 1 Areas where a database can help a core lab Project, experiment and sample tracking Flexibility in experiment design Role based security Automation Custom results
Workshop IIc. Manual interpretation of MS/MS spectra. Ebbing de Jong. Center for Mass Spectrometry and Proteomics Phone (612)625-2280 (612)625-2279
625-2280 (612)625-2279") Workshop IIc Manual interpretation of MS/MS spectra Ebbing de Jong Why MS/MS spectra? The information contained in an MS spectrum (m/z, isotope spacing and therefore z ) is not enough to tell us the amino
Workshop IIc Manual interpretation of MS/MS spectra Ebbing de Jong Why MS/MS spectra? The information contained in an MS spectrum (m/z, isotope spacing and therefore z ) is not enough to tell us the amino
Isobaric Tag based MS Quantification Algorithms Analysis and Implementation
 Isobaric Tag based MS Quantification Algorithms Analysis and Implementation Master s degree in Proteomics and Bioinformatics Written by Sankar Martial Supervisors: Nicolas Budin 1, Pierre-Alain Binz 1
Isobaric Tag based MS Quantification Algorithms Analysis and Implementation Master s degree in Proteomics and Bioinformatics Written by Sankar Martial Supervisors: Nicolas Budin 1, Pierre-Alain Binz 1
Mass Spectrometry Signal Calibration for Protein Quantitation
 Cambridge Isotope Laboratories, Inc. www.isotope.com Proteomics Mass Spectrometry Signal Calibration for Protein Quantitation Michael J. MacCoss, PhD Associate Professor of Genome Sciences University of
Cambridge Isotope Laboratories, Inc. www.isotope.com Proteomics Mass Spectrometry Signal Calibration for Protein Quantitation Michael J. MacCoss, PhD Associate Professor of Genome Sciences University of
ID of alternative translational initiation events. Description of gene function Reference of NCBI database access and relative literatures
 Data resource: In this database, 650 alternatively translated variants assigned to a total of 300 genes are contained. These database records of alternative translational initiation have been collected
Data resource: In this database, 650 alternatively translated variants assigned to a total of 300 genes are contained. These database records of alternative translational initiation have been collected
Interpretation of MS-Based Proteomics Data
 Interpretation of MS-Based Proteomics Data Yet-Ran Chen, 陳 逸 然 Agricultural Biotechnology Research Center Academia Sinica Brief Overview of Protein Identification Workflow Protein Sample Specific Protein
Interpretation of MS-Based Proteomics Data Yet-Ran Chen, 陳 逸 然 Agricultural Biotechnology Research Center Academia Sinica Brief Overview of Protein Identification Workflow Protein Sample Specific Protein
Quan%ta%ve proteomics. Maarten Altelaar, 2014
 Quan%ta%ve proteomics Maarten Altelaar, 2014 Proteomics Altelaar et al. Nat Rev Gen 14, 2013, 35-48 Quan%ta%ve proteomics Quan%ta%ve proteomics Control Diseased, s%mulated, Knock down, etc. How quan%ta%ve
Quan%ta%ve proteomics Maarten Altelaar, 2014 Proteomics Altelaar et al. Nat Rev Gen 14, 2013, 35-48 Quan%ta%ve proteomics Quan%ta%ve proteomics Control Diseased, s%mulated, Knock down, etc. How quan%ta%ve
Advantages of the LTQ Orbitrap for Protein Identification in Complex Digests
 Application Note: 386 Advantages of the LTQ Orbitrap for Protein Identification in Complex Digests Rosa Viner, Terry Zhang, Scott Peterman, and Vlad Zabrouskov, Thermo Fisher Scientific, San Jose, CA,
Application Note: 386 Advantages of the LTQ Orbitrap for Protein Identification in Complex Digests Rosa Viner, Terry Zhang, Scott Peterman, and Vlad Zabrouskov, Thermo Fisher Scientific, San Jose, CA,
Pep-Miner: A Novel Technology for Mass Spectrometry-Based Proteomics
 Pep-Miner: A Novel Technology for Mass Spectrometry-Based Proteomics Ilan Beer Haifa Research Lab Dec 10, 2002 Pep-Miner s Location in the Life Sciences World The post-genome era - the age of proteome
Pep-Miner: A Novel Technology for Mass Spectrometry-Based Proteomics Ilan Beer Haifa Research Lab Dec 10, 2002 Pep-Miner s Location in the Life Sciences World The post-genome era - the age of proteome
Searching Nucleotide Databases
 Searching Nucleotide Databases 1 When we search a nucleic acid databases, Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from the forward strand and 3 reading frames
Searching Nucleotide Databases 1 When we search a nucleic acid databases, Mascot always performs a 6 frame translation on the fly. That is, 3 reading frames from the forward strand and 3 reading frames
Mass Spectra Alignments and their Significance
 Mass Spectra Alignments and their Significance Sebastian Böcker 1, Hans-Michael altenbach 2 1 Technische Fakultät, Universität Bielefeld 2 NRW Int l Graduate School in Bioinformatics and Genome Research,
Mass Spectra Alignments and their Significance Sebastian Böcker 1, Hans-Michael altenbach 2 1 Technische Fakultät, Universität Bielefeld 2 NRW Int l Graduate School in Bioinformatics and Genome Research,
La Protéomique : Etat de l art et perspectives
 La Protéomique : Etat de l art et perspectives Odile Schiltz Institut de Pharmacologie et de Biologie Structurale CNRS, Université de Toulouse, Odile.Schiltz@ipbs.fr Protéomique et Spectrométrie de Masse
La Protéomique : Etat de l art et perspectives Odile Schiltz Institut de Pharmacologie et de Biologie Structurale CNRS, Université de Toulouse, Odile.Schiltz@ipbs.fr Protéomique et Spectrométrie de Masse
Metabolomics Software Tools. Xiuxia Du, Paul Benton, Stephen Barnes
 Metabolomics Software Tools Xiuxia Du, Paul Benton, Stephen Barnes Outline 2 Introduction Software Tools for LC-MS metabolomics Software Tools for GC-MS metabolomics Software Tools for Statistical Analysis
Metabolomics Software Tools Xiuxia Du, Paul Benton, Stephen Barnes Outline 2 Introduction Software Tools for LC-MS metabolomics Software Tools for GC-MS metabolomics Software Tools for Statistical Analysis
ms-data-core-api: An open-source, metadata-oriented library for computational proteomics
 Application Note ms-data-core-api: An open-source, metadata-oriented library for computational proteomics Yasset Perez-Riverol a, Julian Uszkoreit b, Aniel Sanchez c, Tobias Ternent a, Noemi del Toro a,
Application Note ms-data-core-api: An open-source, metadata-oriented library for computational proteomics Yasset Perez-Riverol a, Julian Uszkoreit b, Aniel Sanchez c, Tobias Ternent a, Noemi del Toro a,
Pinpointing phosphorylation sites using Selected Reaction Monitoring and Skyline
 Pinpointing phosphorylation sites using Selected Reaction Monitoring and Skyline Christina Ludwig group of Ruedi Aebersold, ETH Zürich The challenge of phosphosite assignment Peptides Phosphopeptides MS/MS
Pinpointing phosphorylation sites using Selected Reaction Monitoring and Skyline Christina Ludwig group of Ruedi Aebersold, ETH Zürich The challenge of phosphosite assignment Peptides Phosphopeptides MS/MS
Retrospective Analysis of a Host Cell Protein Perfect Storm: Identifying Immunogenic Proteins and Fixing the Problem
 Retrospective Analysis of a Host Cell Protein Perfect Storm: Identifying Immunogenic Proteins and Fixing the Problem Kevin Van Cott, Associate Professor Dept. of Chemical and Biomolecular Engineering Nebraska
Retrospective Analysis of a Host Cell Protein Perfect Storm: Identifying Immunogenic Proteins and Fixing the Problem Kevin Van Cott, Associate Professor Dept. of Chemical and Biomolecular Engineering Nebraska
Mascot Search Results FAQ
 Mascot Search Results FAQ 1 We had a presentation with this same title at our 2005 user meeting. So much has changed in the last 6 years that it seemed like a good idea to re-visit the topic. Just about
Mascot Search Results FAQ 1 We had a presentation with this same title at our 2005 user meeting. So much has changed in the last 6 years that it seemed like a good idea to re-visit the topic. Just about
A Streamlined Workflow for Untargeted Metabolomics
 A Streamlined Workflow for Untargeted Metabolomics Employing XCMS plus, a Simultaneous Data Processing and Metabolite Identification Software Package for Rapid Untargeted Metabolite Screening Baljit K.
A Streamlined Workflow for Untargeted Metabolomics Employing XCMS plus, a Simultaneous Data Processing and Metabolite Identification Software Package for Rapid Untargeted Metabolite Screening Baljit K.
The Open2Dprot Proteomics Project for n-dimensional Protein Expression Data Analysis
 The Open2Dprot Proteomics Project for n-dimensional Protein Expression Data Analysis http://open2dprot.sourceforge.net/ Revised 2-05-2006 * (cf. 2D-LC) Introduction There is a need for integrated proteomics
The Open2Dprot Proteomics Project for n-dimensional Protein Expression Data Analysis http://open2dprot.sourceforge.net/ Revised 2-05-2006 * (cf. 2D-LC) Introduction There is a need for integrated proteomics
Chapter 14. Modeling Experimental Design for Proteomics. Jan Eriksson and David Fenyö. Abstract. 1. Introduction
 Chapter Modeling Experimental Design for Proteomics Jan Eriksson and David Fenyö Abstract The complexity of proteomes makes good experimental design essential for their successful investigation. Here,
Chapter Modeling Experimental Design for Proteomics Jan Eriksson and David Fenyö Abstract The complexity of proteomes makes good experimental design essential for their successful investigation. Here,
Thermo Scientific PepFinder Software A New Paradigm for Peptide Mapping
 Thermo Scientific PepFinder Software A New Paradigm for Peptide Mapping For Conclusive Characterization of Biologics Deep Protein Characterization Is Crucial Pharmaceuticals have historically been small
Thermo Scientific PepFinder Software A New Paradigm for Peptide Mapping For Conclusive Characterization of Biologics Deep Protein Characterization Is Crucial Pharmaceuticals have historically been small
High Throughput Proteomics
 High Throughput Proteomics 1 High throughput means... Continuous real-time searching of data Human scrutiny of results is not practical High throughput doesn t necessarily mean large scale. We would characterise
High Throughput Proteomics 1 High throughput means... Continuous real-time searching of data Human scrutiny of results is not practical High throughput doesn t necessarily mean large scale. We would characterise
Accurate Mass Screening Workflows for the Analysis of Novel Psychoactive Substances
 Accurate Mass Screening Workflows for the Analysis of Novel Psychoactive Substances TripleTOF 5600 + LC/MS/MS System with MasterView Software Adrian M. Taylor AB Sciex Concord, Ontario (Canada) Overview
Accurate Mass Screening Workflows for the Analysis of Novel Psychoactive Substances TripleTOF 5600 + LC/MS/MS System with MasterView Software Adrian M. Taylor AB Sciex Concord, Ontario (Canada) Overview
Tutorial for proteome data analysis using the Perseus software platform
 Tutorial for proteome data analysis using the Perseus software platform Laboratory of Mass Spectrometry, LNBio, CNPEM Tutorial version 1.0, January 2014. Note: This tutorial was written based on the information
Tutorial for proteome data analysis using the Perseus software platform Laboratory of Mass Spectrometry, LNBio, CNPEM Tutorial version 1.0, January 2014. Note: This tutorial was written based on the information
Electrospray Ion Trap Mass Spectrometry. Introduction
 Electrospray Ion Source Electrospray Ion Trap Mass Spectrometry Introduction The key to using MS for solutions is the ability to transfer your analytes into the vacuum of the mass spectrometer as ionic
Electrospray Ion Source Electrospray Ion Trap Mass Spectrometry Introduction The key to using MS for solutions is the ability to transfer your analytes into the vacuum of the mass spectrometer as ionic
PEAKS Studio User Manual (v5.3) PEAKS Team
 PEAKS Team") PEAKS Studio User Manual (v5.3) PEAKS Team PEAKS Studio User Manual (v5.3) PEAKS Team Publication date 2011 Table of Contents I. Basic Operations... 1 1. Welcome to PEAKS... 4 1. Main Functions... 4 2.
PEAKS Studio User Manual (v5.3) PEAKS Team PEAKS Studio User Manual (v5.3) PEAKS Team Publication date 2011 Table of Contents I. Basic Operations... 1 1. Welcome to PEAKS... 4 1. Main Functions... 4 2.
Data mining with Mascot Integra ASMS 2005
 Data mining with Mascot Integra 1 What is Mascot Integra? Fully functional out-the-box solution for proteomics workflow and data management Support for all the major mass-spectrometry data systems Powered
Data mining with Mascot Integra 1 What is Mascot Integra? Fully functional out-the-box solution for proteomics workflow and data management Support for all the major mass-spectrometry data systems Powered
When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want
 1 When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want to search other databases as well. There are very
1 When you install Mascot, it includes a copy of the Swiss-Prot protein database. However, it is almost certain that you and your colleagues will want to search other databases as well. There are very
Introduction to Proteomics
 Introduction to Proteomics Why Proteomics? Same Genome Different Proteome Black Swallowtail - larvae and butterfly Biological Complexity Yeast - a simple proteome 6,113 proteins = 344,855 tryptic peptides
Introduction to Proteomics Why Proteomics? Same Genome Different Proteome Black Swallowtail - larvae and butterfly Biological Complexity Yeast - a simple proteome 6,113 proteins = 344,855 tryptic peptides
Mass Frontier Version 7.0
 Mass Frontier Version 7.0 User Guide XCALI-97349 Revision A February 2011 2011 Thermo Fisher Scientific Inc. All rights reserved. Mass Frontier, Mass Frontier Server Manager, Fragmentation Library, Spectral
Mass Frontier Version 7.0 User Guide XCALI-97349 Revision A February 2011 2011 Thermo Fisher Scientific Inc. All rights reserved. Mass Frontier, Mass Frontier Server Manager, Fragmentation Library, Spectral
SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays
 Protocol SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878 Order per fax: +49-30-6392-7888 or e-mail: www: peptide@jpt.com www.jpt.com
Protocol SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878 Order per fax: +49-30-6392-7888 or e-mail: www: peptide@jpt.com www.jpt.com
MaxQuant User s Guide Version 1.2.2.5
 MaxQuant User s Guide Version 1.2.2.5 Jűrgen Cox and Matthias Mann Nature Biotechnology 26, 1367-1372 (2008) Sara ten Have 2012 http://www.lamondlab.com/ http://greproteomics.lifesci.dundee.ac.uk/ References
MaxQuant User s Guide Version 1.2.2.5 Jűrgen Cox and Matthias Mann Nature Biotechnology 26, 1367-1372 (2008) Sara ten Have 2012 http://www.lamondlab.com/ http://greproteomics.lifesci.dundee.ac.uk/ References
Statistical Analysis Strategies for Shotgun Proteomics Data
 Statistical Analysis Strategies for Shotgun Proteomics Data Ming Li, Ph.D. Cancer Biostatistics Center Vanderbilt University Medical Center Ayers Institute Biomarker Pipeline normal shotgun proteome analysis
Statistical Analysis Strategies for Shotgun Proteomics Data Ming Li, Ph.D. Cancer Biostatistics Center Vanderbilt University Medical Center Ayers Institute Biomarker Pipeline normal shotgun proteome analysis
Building innovative drug discovery alliances. Evotec Munich. Quantitative Proteomics to Support the Discovery & Development of Targeted Drugs
 Building innovative drug discovery alliances Evotec Munich Quantitative Proteomics to Support the Discovery & Development of Targeted Drugs Evotec AG, Evotec Munich, June 2013 About Evotec Munich A leader
Building innovative drug discovery alliances Evotec Munich Quantitative Proteomics to Support the Discovery & Development of Targeted Drugs Evotec AG, Evotec Munich, June 2013 About Evotec Munich A leader
Integrated Data Mining Strategy for Effective Metabolomic Data Analysis
 The First International Symposium on Optimization and Systems Biology (OSB 07) Beijing, China, August 8 10, 2007 Copyright 2007 ORSC & APORC pp. 45 51 Integrated Data Mining Strategy for Effective Metabolomic
The First International Symposium on Optimization and Systems Biology (OSB 07) Beijing, China, August 8 10, 2007 Copyright 2007 ORSC & APORC pp. 45 51 Integrated Data Mining Strategy for Effective Metabolomic
Application Note # MT-90 MALDI-TDS: A Coherent MALDI Top-Down-Sequencing Approach Applied to the ABRF-Protein Research Group Study 2008
 Bruker Daltonics Application Note # MT-90 MALDI-TDS: A Coherent MALDI Top-Down-Sequencing Approach Applied to the ABRF-Protein Research Group Study 2008 In the ABRF-PRG study 2008 [*] the ability to characterize
Bruker Daltonics Application Note # MT-90 MALDI-TDS: A Coherent MALDI Top-Down-Sequencing Approach Applied to the ABRF-Protein Research Group Study 2008 In the ABRF-PRG study 2008 [*] the ability to characterize
Preprocessing, Management, and Analysis of Mass Spectrometry Proteomics Data
 Preprocessing, Management, and Analysis of Mass Spectrometry Proteomics Data M. Cannataro, P. H. Guzzi, T. Mazza, and P. Veltri Università Magna Græcia di Catanzaro, Italy 1 Introduction Mass Spectrometry
Preprocessing, Management, and Analysis of Mass Spectrometry Proteomics Data M. Cannataro, P. H. Guzzi, T. Mazza, and P. Veltri Università Magna Græcia di Catanzaro, Italy 1 Introduction Mass Spectrometry
Shotgun Proteomic Analysis. Department of Cell Biology The Scripps Research Institute
 Shotgun Proteomic Analysis Department of Cell Biology The Scripps Research Institute Biological/Functional Resolution of Experiments Organelle Multiprotein Complex Cells/Tissues Function Expression Analysis
Shotgun Proteomic Analysis Department of Cell Biology The Scripps Research Institute Biological/Functional Resolution of Experiments Organelle Multiprotein Complex Cells/Tissues Function Expression Analysis
Unique Software Tools to Enable Quick Screening and Identification of Residues and Contaminants in Food Samples using Accurate Mass LC-MS/MS
 Unique Software Tools to Enable Quick Screening and Identification of Residues and Contaminants in Food Samples using Accurate Mass LC-MS/MS Using PeakView Software with the XIC Manager to Get the Answers
Unique Software Tools to Enable Quick Screening and Identification of Residues and Contaminants in Food Samples using Accurate Mass LC-MS/MS Using PeakView Software with the XIC Manager to Get the Answers
Pesticide Analysis by Mass Spectrometry
 Pesticide Analysis by Mass Spectrometry Purpose: The purpose of this assignment is to introduce concepts of mass spectrometry (MS) as they pertain to the qualitative and quantitative analysis of organochlorine
Pesticide Analysis by Mass Spectrometry Purpose: The purpose of this assignment is to introduce concepts of mass spectrometry (MS) as they pertain to the qualitative and quantitative analysis of organochlorine
Quantitative mass spectrometry in proteomics: a critical review
 Anal Bioanal Chem (2007) 389:1017 1031 DOI 10.1007/s00216-007-1486-6 REVIEW Quantitative mass spectrometry in proteomics: a critical review Marcus Bantscheff & Markus Schirle & Gavain Sweetman & Jens Rick
Anal Bioanal Chem (2007) 389:1017 1031 DOI 10.1007/s00216-007-1486-6 REVIEW Quantitative mass spectrometry in proteomics: a critical review Marcus Bantscheff & Markus Schirle & Gavain Sweetman & Jens Rick
A Tool To Visualize and Evaluate Data Obtained by Liquid Chromatography-Electrospray Ionization-Mass Spectrometry
 Anal. Chem. 2004, 76, 3856-3860 A Tool To Visualize and Evaluate Data Obtained by Liquid Chromatography-Electrospray Ionization-Mass Spectrometry Xiao-jun Li,* Patrick G. A. Pedrioli, Jimmy Eng, Dan Martin,
Anal. Chem. 2004, 76, 3856-3860 A Tool To Visualize and Evaluate Data Obtained by Liquid Chromatography-Electrospray Ionization-Mass Spectrometry Xiao-jun Li,* Patrick G. A. Pedrioli, Jimmy Eng, Dan Martin,
Mass spectrometry-based proteomics in biomedical research: emerging technologies and future strategies
 Mass spectrometry-based proteomics in biomedical research: emerging technologies and future strategies Geraldine M. Walsh 1,2, Jason C. Rogalski 1,2, Cordula Klockenbusch 1 and Juergen Kast 1,2,3, * In
Mass spectrometry-based proteomics in biomedical research: emerging technologies and future strategies Geraldine M. Walsh 1,2, Jason C. Rogalski 1,2, Cordula Klockenbusch 1 and Juergen Kast 1,2,3, * In
Management of Proteomics Data: 2D Gel Electrophoresis and Other Methods
 Management of Proteomics Data: 2D Gel Electrophoresis and Other Methods Philip Andrews National Resource for Proteomics & Pathway Mapping Michigan Proteome Consortium University of Michigan Outline of
Management of Proteomics Data: 2D Gel Electrophoresis and Other Methods Philip Andrews National Resource for Proteomics & Pathway Mapping Michigan Proteome Consortium University of Michigan Outline of
MassHunter for Agilent GC/MS & GC/MS/MS
 MassHunter for Agilent GC/MS & GC/MS/MS Next Generation Data Analysis Software Presented by : Terry Harper GC/MS Product Specialist 1 Outline of Topics Topic 1: Introduction to MassHunter Topic 2: Data
MassHunter for Agilent GC/MS & GC/MS/MS Next Generation Data Analysis Software Presented by : Terry Harper GC/MS Product Specialist 1 Outline of Topics Topic 1: Introduction to MassHunter Topic 2: Data
Thermo Scientific SIEVE Software for Differential Expression Analysis
 m a s s s p e c t r o m e t r y Thermo Scientific SIEVE Software for Differential Expression Analysis Automated, label-free, semi-quantitative analysis of proteins, peptides, and metabolites based on comparisons
m a s s s p e c t r o m e t r y Thermo Scientific SIEVE Software for Differential Expression Analysis Automated, label-free, semi-quantitative analysis of proteins, peptides, and metabolites based on comparisons
Using Ontologies in Proteus for Modeling Data Mining Analysis of Proteomics Experiments
 Using Ontologies in Proteus for Modeling Data Mining Analysis of Proteomics Experiments Mario Cannataro, Pietro Hiram Guzzi, Tommaso Mazza, and Pierangelo Veltri University Magna Græcia of Catanzaro, 88100
Using Ontologies in Proteus for Modeling Data Mining Analysis of Proteomics Experiments Mario Cannataro, Pietro Hiram Guzzi, Tommaso Mazza, and Pierangelo Veltri University Magna Græcia of Catanzaro, 88100
Thermo Scientific ExactFinder Software
 mass spectrometry Thermo Scientific ExactFinder Software Workflow software for routine targeted and general unknown screening Unifies qualitative and quantitative high-resolution accurate mass workflows
mass spectrometry Thermo Scientific ExactFinder Software Workflow software for routine targeted and general unknown screening Unifies qualitative and quantitative high-resolution accurate mass workflows
Identification of Serum Protein Biomarkers for Autistic Spectrum Disorder. Melissa Butkiewicz
 Identification of Serum Protein Biomarkers for Autistic Spectrum Disorder by Melissa Butkiewicz Clarkson University Identification of Serum Protein Biomarkers for Autistic Spectrum Disorder A Thesis Proposal
Identification of Serum Protein Biomarkers for Autistic Spectrum Disorder by Melissa Butkiewicz Clarkson University Identification of Serum Protein Biomarkers for Autistic Spectrum Disorder A Thesis Proposal
MassMatrix Web Server User Manual
 MassMatrix Web Server User Manual Version 2.2.3 or later Hua Xu, Ph. D. Center for Proteomics & Bioinformatics Case Western Reserve University August 2009 Main Navigation Bar of the Site MassMatrix Web
MassMatrix Web Server User Manual Version 2.2.3 or later Hua Xu, Ph. D. Center for Proteomics & Bioinformatics Case Western Reserve University August 2009 Main Navigation Bar of the Site MassMatrix Web
Software for protein identification and quantitative analysis of mass spectrometry data used for protein characterization and proteomics Version 5.
 Release Notes ProteinPilot Software for protein identification and quantitative analysis of mass spectrometry data used for protein characterization and proteomics Version 5.0 Introduction Thank you for
Release Notes ProteinPilot Software for protein identification and quantitative analysis of mass spectrometry data used for protein characterization and proteomics Version 5.0 Introduction Thank you for
Improving the Metabolite Identification Process with Efficiency and Speed: LightSight Software for Metabolite Identification
 Improving the Metabolite Identification Process with Efficiency and Speed: LightSight Software for Metabolite Identification Overview LightSight Software for Metabolite Identification is a complete environment
Improving the Metabolite Identification Process with Efficiency and Speed: LightSight Software for Metabolite Identification Overview LightSight Software for Metabolite Identification is a complete environment
MarkerView Software 1.2.1 for Metabolomic and Biomarker Profiling Analysis
 MarkerView Software 1.2.1 for Metabolomic and Biomarker Profiling Analysis Overview MarkerView software is a novel program designed for metabolomics applications and biomarker profiling workflows 1. Using
MarkerView Software 1.2.1 for Metabolomic and Biomarker Profiling Analysis Overview MarkerView software is a novel program designed for metabolomics applications and biomarker profiling workflows 1. Using
Definition of the Measurand: CRP
 A Reference Measurement System for C-reactive Protein David M. Bunk, Ph.D. Chemical Science and Technology Laboratory National Institute of Standards and Technology Definition of the Measurand: Human C-reactive
A Reference Measurement System for C-reactive Protein David M. Bunk, Ph.D. Chemical Science and Technology Laboratory National Institute of Standards and Technology Definition of the Measurand: Human C-reactive
ALGORITHMS FOR SHOTGUN PROTEOMICS SPECTRAL IDENTIFICATION AND QUALITY ASSESSMENT. Ze-Qiang Ma
 ALGORITHMS FOR SHOTGUN PROTEOMICS SPECTRAL IDENTIFICATION AND QUALITY ASSESSMENT By Ze-Qiang Ma Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment
ALGORITHMS FOR SHOTGUN PROTEOMICS SPECTRAL IDENTIFICATION AND QUALITY ASSESSMENT By Ze-Qiang Ma Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment
Comparative LC-MS: A landscape of peaks and valleys
 Proteomics 2008, 8, 731 749 DOI 10.1002/pmic.200700694 731 REVIEW Comparative LC-MS: A landscape of peaks and valleys Antoine H. P. America and Jan H. G. Cordewener Plant Research International, Wageningen
Proteomics 2008, 8, 731 749 DOI 10.1002/pmic.200700694 731 REVIEW Comparative LC-MS: A landscape of peaks and valleys Antoine H. P. America and Jan H. G. Cordewener Plant Research International, Wageningen
Automated reporting from gel-based proteomics experiments using the open source Proteios database application
 668 DOI 10.1002/pmic.200600814 Proteomics 2007, 7, 668 674 RESEARCH ARTICLE Automated reporting from gel-based proteomics experiments using the open source Proteios database application Fredrik Levander
668 DOI 10.1002/pmic.200600814 Proteomics 2007, 7, 668 674 RESEARCH ARTICLE Automated reporting from gel-based proteomics experiments using the open source Proteios database application Fredrik Levander