HYDROGEN EXCHANGE MASS SPECTROMETRY FOR THE ANALYSIS OF PROTEIN DYNAMICS
|
|
|
- Lee Brown
- 8 years ago
- Views:
Transcription
1 HYDROGEN EXCHANGE MASS SPECTROMETRY FOR THE ANALYSIS OF PROTEIN DYNAMICS Thomas E. Wales and John R. Engen* Department of Chemistry, University of New Mexico, Albuquerque, New Mexico Received 5 April 2005; received (revised) 5 July 2005; accepted 6 July 2005 Published online 5 October 2005 in Wiley InterScience ( DOI /mas Hydrogen exchange coupled to mass spectrometry (MS) has become a valuable analytical tool for the study of protein dynamics. By combining information about protein dynamics with more classical functional data, a more thorough understanding of protein function can be obtained. In many cases, protein dynamics are directly related to specific protein functions such as conformational changes during enzyme activation or protein movements during binding. The method is made possible because labile backbone hydrogens in a protein will exchange with deuterium atoms when the protein is placed in a D 2 O solution. The subsequent increase in protein mass over time is measured with high-resolution MS. The location of the deuterium incorporation is determined by monitoring deuterium incorporation in peptic fragments that are produced after the labeling reaction. In this review, we will summarize the general principles of the method, discuss the latest variations on the experimental protocol that probe different types of protein movements, and review other recent work and improvements in the field. # 2005 Wiley Periodicals, Inc., Mass Spec Rev 25: , 2006 Keywords: deuterium; electrospray; MALDI; protein folding; conformation I. INTRODUCTION Proteins are not static structures in solution. They move and flex naturally or in response to external stimuli. The movements of proteins, collectively termed protein dynamics, can be extremely important for protein function. To ascertain how dynamics play a role in protein function, actual protein motions themselves need to be investigated and understood. Currently, there are not a large number of analytical tools capable of providing the necessary resolution to connect specific protein movements with protein function. The development of new tools and refinement of those that are currently in use is therefore of great interest. To this end, hydrogen exchange (HX) coupled with mass spectrometry (MS) presents an opportunity for the analysis of proteins and protein motions in ways that were not imagined even 5 years ago. Traditionally, hydrogen exchange methodology has been used in conjunction with NMR analysis [see (Dyson & Contract grant sponsor: The National Institutes of Health; Contract grant numbers: R01-GM070590, R01-GM068901, R24-CA088339, P20-RR *Correspondence to: John R. Engen, Clark Hall 242, MSC , Department of Chemistry, University of New Mexico, Albuquerque, NM engen@unm.edu Wright, 2004) for a review]. In comparison, hydrogen exchange MS is a more recent development. The first demonstrated use of HX MS came shortly after the development of electrospray ionization (Chowdhury, Katta, & Chait, 1990; Katta & Chait, 1991). Important developments through the 1990s were led by Prof. David Smith [reviewed in (Smith, Deng, & Zhang, 1997; Engen & Smith, 2001)]. Hydrogen exchange MS has been the subject of several comprehensive reviews (Kaltashov & Eyles, 2002a,b; Hoofnagle, Resing, & Ahn, 2003; Eyles & Kaltashov, 2004; Garcia, Pantazatos, & Villarreal, 2004). Recent developments in this field, which will be summarized in the following sections, offer unparalleled limits of detection, low sample consumption requirements, the promise of single amino acid resolution, potential for automation and the ability to analyze increasingly more complex mixtures. Future refinements that could substantially improve the method will also be discussed. II. OVERVIEW OF THE METHOD An general scheme for hydrogen exchange MS experiments is shown in Figure 1. The integration of deuterium into the protein(s) of interest relies on the natural phenomena of hydrogen exchange. The introduction of deuterium into a peptide or protein can be accomplished in several ways and its incorporation can be analyzed using several methods. The mass spectrometer is used to monitor the increase in mass as hydrogen is exchanged for deuterium. Throughout this review, we discuss several parts in the scheme (Fig. 1, numbered circles) where recent improvements have been made and where future refinements are anticipated. III. HYDROGEN EXCHANGE FUNDAMENTALS The details of hydrogen exchange mechanisms have been widely reviewed (Hvidt & Nielsen, 1966; Woodward, Simon, & Tüchsen, 1982; Englander & Kallenbach, 1984; Kim & Woodward, 1993; Mayo & Baldwin, 1993; Bai et al., 1995; Miller & Dill, 1995; Loh et al., 1996; Clarke, Itzhaki, & Fersht, 1997; Kaltashov & Eyles, 2002b; Hoofnagle, Resing, & Ahn, 2003; Eyles & Kaltashov, 2004; Krishna et al., 2004; Smith, Deng, & Zhang, 1997). Here a short summary, collected from these references, on the basics of hydrogen exchange and how it is applied to the study of protein dynamics is presented. Hydrogens that are located at peptide amide linkages (also referred to as the backbone amide hydrogens) undergo replacement with deuterons within 1 10 s when the peptides are incubated in D 2 O Mass Spectrometry Reviews, 2006, 25, # 2005 by Wiley Periodicals, Inc.

2 PROTEIN DYNAMICS BY HYDROGEN EXCHANGE MASS SPECTROMETRY & at pd 7.0. In folded proteins, some backbone amide hydrogens exchange quickly while others exchange only after months. The rates of the most slowly exchanging amide hydrogens may be reduced by as much as 10 8 of their rates in unfolded forms of the same protein (Englander & Kallenbach, 1984). Nearly all peptide amide hydrogens in folded proteins are hydrogen bonded, either intramolecularly to another part of the protein or to water. The large reduction of amide hydrogen exchange rates in folded proteins is primarily due to restricted access of solvent to the interior of the protein and to intramolecular hydrogen bonding. It is not possible to differentiate between these two contributing parameters as they occur concomitantly. At physiological ph, base-catalyzed exchange is the dominant mechanism for hydrogen exchange. Base-catalyzed isotope exchange can occur only when a hydrogen bond is severed in the presence of the catalyst (hydroxide) and the source of the new hydrogen (water). The rate constant for isotope exchange at each individual amide linkage in a normally folded protein, k ex, can be described by Equation 1 k ex ¼ k f þ k u ¼ðb þ K unf Þk 2 ð1þ where k ex is expressed as the sum of the contributions of exchange from folded (k f ) and unfolded (k u ) forms of the protein (Kim & Woodward, 1993). The mechanisms described by Equation 1 are illustrated graphically in Figure 2. Exchange from the folded form likely dominates for amide hydrogens that are not participating in intramolecular hydrogen bonding and that are located near the surface. Exchange in unfolded forms requires substantial movement of the backbone. Unfolding to expose backbone amide sites to deuterium can be isolated to small regions (localized unfolding) or may involve the entire protein (global unfolding). The rate constant for exchange from the folded state, k f,is described by Equation 2 k f ¼ bk 2 where b is a probability factor for exchange from folded forms and k 2 is the rate constant for HX at each amide linkage in an unstructured peptide, a value that can be calculated (Bai et al., 1993). The value for b ranges from 0 to 1 and is a function of several parameters including solvent accessibility and intramolecular hydrogen bonding. When b is closer to 1, there is a higher probability that a particular amide hydrogen is exposed to water and catalyst at the same time that it is also exchange competent. K unf in Equation 1 is the equilibrium constant describing the unfolding process. HX NMR studies have used denaturants to distinguish between b and K unf (Bai et al., 1994; Itzhaki, Neira, & Fersht, 1997; Chamberlain & Marqusee, 1998). The rate constant for exchange from unfolded forms of proteins depends on the rate constant for exchange from an unfolded peptide (k 2 ) as well as the unfolding dynamics described by k 1 and k 1 as shown in Equation 3 where F and U are the folded and unfolded forms, respectively. ð2þ F H Ð k1 U H! k2 U k 1 D Ð F D ð3þ k 1 D 2 O k 1 When k 2 >> k 1 (termed EX1 kinetics), the rate constant for exchange from unfolded forms is given by the unfolding rate constant k 1 (Eq. 4). k u ¼ k 1 ð4þ However, under physiological conditions it is more common for k 1 >> k 2. In this case (EX2 kinetics), the rate constant for exchange from unfolded forms is given by Equation 5 where K unf is the equilibrium constant describing the unfolding process. k u ¼ k 1 k 2 ¼ K unf k 2 ð5þ k 1 While only a few proteins undergo EX1 kinetics naturally, all proteins undergo EX2 exchange kinetics under physiological conditions. EX2 kinetics may be envisioned as involving many rapid and random visits to a state capable of exchange. However, the probability of exchange during a single visit is small. EX1 kinetics are described as a cooperative unfolding event involving several residues, all of which exchange before refolding (k 1 ) occurs (Eq. 3). Proteins can be induced to exhibit EX1 kinetics with denaturant (Deng & Smith, 1998) or by increasing ph (Swint-Kruse & Robertson, 1996). Some proteins may contain regions that undergo EX1 and EX2 kinetics simultaneously. Regions in which exchange occurs by either EX1 or EX2 kinetics can be identified by characteristic isotope patterns in mass spectra (Miranker et al., 1993). Structural changes required for HX described by k f and k u differ in the magnitude of atomic displacement(s) required for isotope exchange. Due to the highly compact nature of proteins in their native state, relative to their denatured states, exchange at individual sites is believed to involve small atomic movements, probably less than an angstrom, but sufficient to allow diffusion of OD and D 2 O to the exchange site (Kim & Woodward, 1993). In parallel with this highly localized motion, short segments, as well as the entire backbone of a protein, can exchange through unfolding processes. Molecular motions associated with unfolding of large segments of the backbone require displacing many atoms several angstroms from their equilibrium positions in the native structure and global unfolding requires gross movement of the entire backbone. Thus, exchange from the folded form (k f )of a protein involves primarily low amplitude motions (small displacement) while exchange from unfolded forms (k u ) requires much larger amplitude motions. Results of a theoretical study by Miller and Dill suggest that large structural changes with little changes in free energy are possible, but uncommon (Miller & Dill, 1995). IV. DEUTERIUM INTRODUCTION: DEUTERIUM, MEET PROTEIN; PROTEIN, MEET DEUTERIUM To measure the incorporation of deuterium into a protein, the protein must, of course, be exposed to deuterium. The introduction of deuterium sounds like a relatively simple process. However, different methods of deuterium introduction allow one to probe different aspects of protein dynamics. In addition, the sample quantity requirements of the mass spectrometer, 159

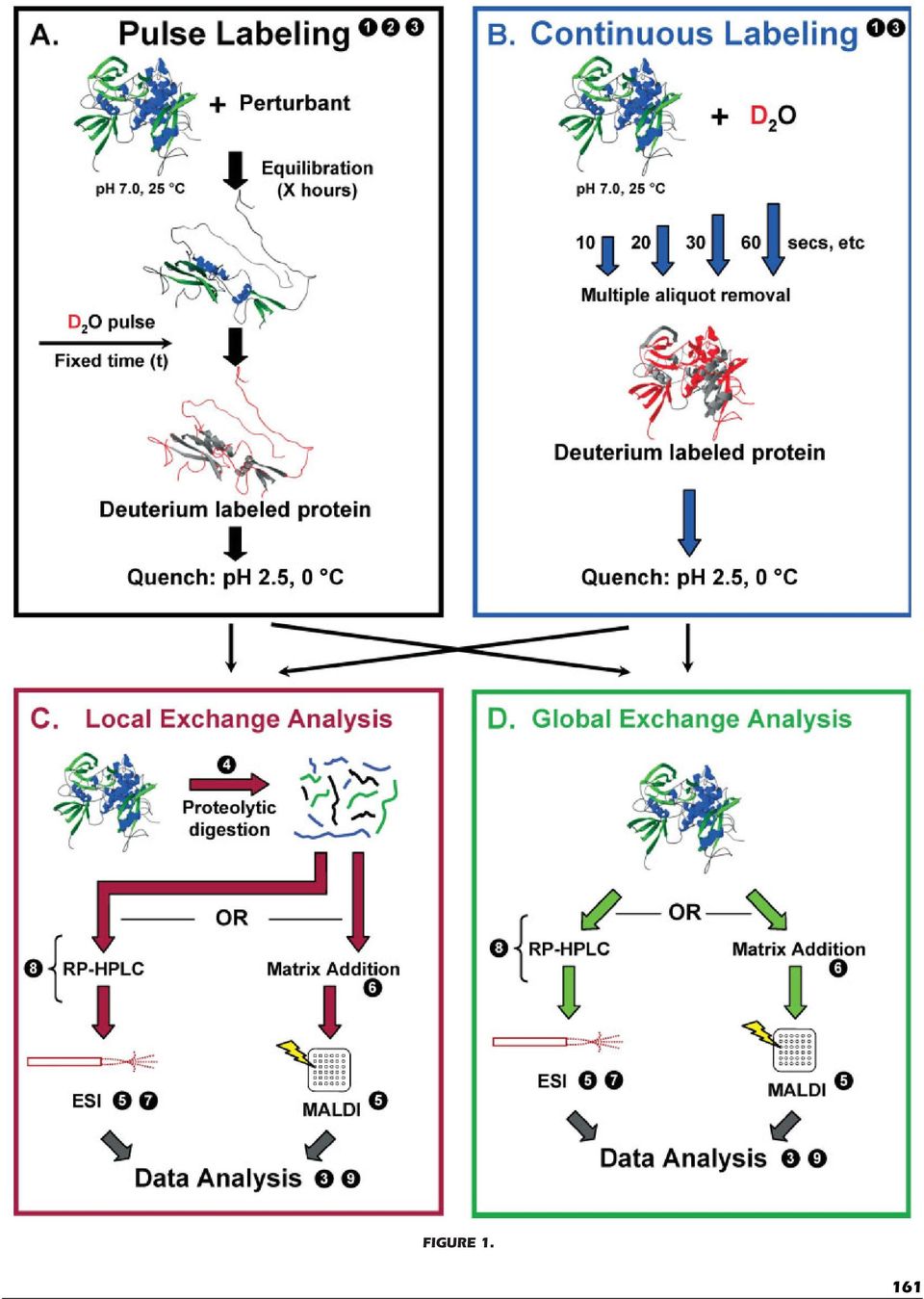
3 & WALES AND ENGEN especially in the case of analysis of protein complexes, may dictate D 2 O introduction of a specific type. The primary method for introducing deuterium into a protein sample is by dilution. Typically, a solution of protein in a protiated buffer is diluted with a deuterated buffer that has a deuterium content of 99% or more. Dilutions of 15-fold or greater will produce final deuterium concentrations of >95%. This serves to force the labeling reaction (k 2 ) in one direction (see Eq. 3). However, with this labeling method the original protein sample is diluted. Such a dilution may not be compatible with the sample quantity requirements of the mass spectrometer and such diluted samples may therefore require an additional experimental step to concentrate the protein sample prior to analysis. Concentration can be accomplished by rapidly trapping the protein with online HPLC at higher flow rates (usually >100 ml/min). The concentration step works best when analyses are performed with online HPLC-ESI as too much deuterium would be lost if this step were done prior to MALDI mass analyses (see below). An alternative to the dilution technique is to carry out a rapid buffer switch with small gel filtration spin columns (Engen & Smith, 2000). Although the buffer switch technique takes a bit longer than the dilution method, the protein is not nearly as diluted. There are two kinds of labeling experiments: continuous labeling and pulse labeling [see also (Deng, Pan, & Smith, 1999a)]. In continuous labeling experiments (Fig. 1, top right), protein is exposed to D 2 O while the populations of folded and unfolded species are in flux. The populations may be in flux as a result of natural protein motions that result from some population-altering force (i.e. the addition of denaturant, change in ph or temperature, in response to protein function, ligand binding, or protein protein complex formation, etc.) designed to cause a shift in the population of folded versus unfolded (or the reverse depending on the experiment). Once a protein has made a transition from a folded to unfolded state, it becomes labeled with D 2 O and the mass increases. As the D 2 O concentration is very high, once a molecule is labeled, it is not able to revert to a protiated species (see Eq. 3). In other words, the transition from a protiated species to one that is deuterated is unidirectional. The deuterium level in the protein sample at any point in the course of the labeling experiment integrates the number of molecules in the sample that had unfolded (or folded) up to that point (Miranker et al., 1993). With continuous labeling, it is possible to sample the population of and potentially observe the transition through various intermediate states whose numbers are very small at any given moment and therefore may go undetected using conventional spectroscopic methods. Continuous labeling is most useful for monitoring slow unfolding transitions [i.e. (Engen et al., 1997)], the majority of unfolding events in proteins. Given enough time, all proteins should become totally deuterated during a continuous labeling experiment as a result of protein motions and protein breathing. Because the transition is very slow for many proteins, the addition of denaturants may be used to induce unfolding. Most HX MS experiments involve continuous labeling simply because they are technically simpler to perform. Far fewer experiments are of the pulse labeling variety. Generally in pulse labeling experiments (Fig. 1, top left), a population of protein molecules is either induced to undergo some kind of conformational change by addition of a perturbing agent or it is already in the process of changing its structure through protein folding. The perturbing agent is most often a chemical denaturant, although heat, ph or binding to substrates can also be used. The sample is then exposed to deuterium for a very brief time (the pulse). Only those molecules that are unfolded when the sample is pulse labeled will undergo isotopic exchange; the remainder of the population remains unlabeled. The resulting deuterium levels then indicate the instantaneous population of folded and unfolded molecules. Pulsed labeling has recently been used to identify protein folding mechanisms [see (Wu & Engen, 2004) for a more detailed description] as well as to probe significantly populated kinetic intermediate states in a folding reaction (either on or off pathway intermediates) (Deng & Smith, 1999; Chen et al., 2001; Wintrode et al., 2003; Mazon et al., 2004; Pan & Smith, 2004; Pan et al., 2004; Rojsajjakul et al., 2004). With pulse labeling, it is FIGURE 1. Overall scheme for hydrogen exchange mass spectrometry experiments. A: Pulse labeling. After a protein has been exposed to a perturbant (chemical denaturant, heat, ph, binding, complex formation, pressure, etc.), unfolded regions (gray) become labeled with deuterium (red) during a quick pulse of D 2 O (typically 10 s). Deuterium exchange is quenched by reducing the ph and temperature. B: Continuous labeling. D 2 O buffer is added to a protein (in H 2 O buffer) such that the final D concentration is >95%. After a set period of time, an aliquot of the labeled protein is removed from the original tube and mixed with quench buffer to reduce the ph and temperature. Aliquot removal is repeated for subsequent labeling times. The protein concentration and solution volume are controlled such that all the aliquots are identical upon quench except for the amount of time the protein was exposed to D 2 O. C: Localized exchange information. Quenched samples (from part A, part B, or both) are digested with pepsin or another acid protease. The resulting peptides are analyzed with online HPLC-ESI-MS or with MALDI-MS. The resulting data analysis provides information on deuterium exchange in short fragments of the peptide backbone. D: Global exchange information. Quenched samples (from part A, part B, or both) are directly analyzed with HPLC-ESI-MS or MALDI-MS. The data provide a global picture of how the protein behaves in D 2 O. It is often recommended that Part D be performed prior to Part C. Areas of recent and future improvement have been marked with numbers. 1 Robotic automation of mixing, buffer addition, etc.; 2 Rapid-mixing techniques such as quench-flow analyses (see text for details); 3 Analysis based upon relative deuterium levels instead of absolute levels (see text for details); 4 Use of acid proteases other than porcine pepsin. Alternative proteases include protease type XIII from Aspergillus saitoi and protease type XVIII from Rhizhopus species (Cravello, Lascoux, & Forest, 2003). 5 CID, ECD, and ETD as fragmentation techniques to provide single amino acid resolution of hydrogen exchange information; 6 Solvent-free MALDI; 7 Nano-ESI-MS and other miniaturization; 8 chromatographic and labeling techniques to accomplish HX MS of single proteins in complexes and mixtures; 9 automation of data analysis and increase interpretation speed. 160
 in one direction (see Eq. 3).")
4 FIGURE
5 & WALES AND ENGEN FIGURE 2. Models for hydrogen exchange into the folded form (A) and into unfolded forms (B) of proteins. The unfolding and refolding rate constants are described by k 1 and k 1, respectively. k 2 is the rate constant for exchange from an unfolded peptide, a value that can be calculated (Bai et al., 1993). See text and Equation 3 for further details. possible to complement data obtained using conventional spectroscopic and NMR experiments (Nishimura, Wright, & Dyson, 2003). Pulse labeling experiments may also be performed using a quench-flow scheme originally described for HX MS by (Yang & Smith, 1997) and more recently discussed by Konerman & Simmons, 2003; Wintrode et al., The addition of deuterium and other steps in the preparation of pulse or continuous labeled samples has been automated (see section on Automation later in this review). With automation, the reproducibility of sample preparation is improved. It is anticipated that fully automated sample preparation devices will become commonplace (Fig. 1, circles 1,2). during the HPLC step and for MALDI analyses, losses may occur during the sample preparation process. However, an adjustment can be made to compensate for back-exchange. An adjustment calculation was first described by Zhang & Smith, In the appendix to their 1993 publication, they describe the derivation, accuracy, and proper use of the equation used for the calculation. While other adjustment methods have been described (Resing, Hoofnagle, & Ahn, 1999; Hoofnagle, Resing, & Ahn, 2004), they do not significantly improve upon this original comprehensive description. When properly controlled, the back-exchange in most modern and well tuned ESI mass spectrometers is on the order of 1 3%. An additional 10 20% of the deuterium label may be lost during in-solution digestion and HPLC separation depending on the length of time that it takes to perform each step. However, peptide and protein recovery differ widely [discussed in (Pan & Smith, 2004)]. More losses can be expected when MALDI is used for the analysis [see below and (Kipping & Schierhorn, 2003)]. However, even when 15% of the label reverts back to hydrogen, less than 1 in 6 deuterium is lost. The end result is that if two curves indicating changes in deuterium levels in a given protein or peptide are obtained and show a difference of 2 3 deuterium, a correction for back-exchange will only change the difference between the two curves by Da. Therefore the comparison of exchange curves with and without the correction yields almost no new information except for a closer approximation of the number of deuterium that were incorporated into the protein or peptide of interest. An example of this is shown in Figure 3. Here, the back-exchange correction was applied to raw data where D 2 O losses were abnormally high (35%). After correction for the V. IMPROVING MASS ANALYSIS A. Retaining the Label Once the incorporation of the isotope label is complete, the task becomes the identification of which amides have been deuterated. To maximize the amount of retained label, the deuterium back-exchange to hydrogen must be controlled. Back-exchange is the undesirable exchange of the deuterium for hydrogen and results in loss of some of the label. To minimize back-exchange, sample analysis must be as rapid as possible and be done at 08C. The majority of back-exchange occurs because proteolytic digestion and the subsequent analysis of the deuterium levels are done with protiated solvents. For ESI analyses, losses occur FIGURE 3. Back-exchange correction. A: Back-exchange adjustment equation as described by (Zhang & Smith, 1993). D is the adjusted deuterium level, m is the experimentally observed mass, m 0% is the 0% or undeuterated control, m 100% is the totally deuterated control, and N is the total number of exchangeable amide hydrogens in the sequence of interest. B: Example of the use of the back-exchange correction in part A. The average deuterium loss for this example peptide was 35%. Closed circles: raw data; Open squares: raw data adjusted using the equation in (A); Dashed line: the calculated amount (Bai et al., 1993) of deuterium in an unstructured peptide with the same sequence as the example peptide examined under identical conditions (ph 7.0 and 228C). 162
 and more recently discussed by Konerman & Simmons, 2003; Wintrode et")
6 PROTEIN DYNAMICS BY HYDROGEN EXCHANGE MASS SPECTROMETRY & samples and all other variables related to back-exchange cancel out. FIGURE 4. Model peptide showing the fragmentation sites for b/y and c/z ions [see Roepstorff & Fohlman, 1984]. 35% D 2 O loss, the curve is shifted upward, with the most significant deviation from the raw, uncorrected data occurring at larger deuterium levels. This is an extreme example, as many peptides do not loose 35% of their label during a well controlled experiment. The back-exchange correction is only necessary when one wishes to know the exact number of deuterium atoms in a given protein or peptide fragment. In many cases where biological functional information is being probed, the actual number of deuterons that have exchanged is not as important as where the exchange has occurred. For example, if deuterium levels are being compared in peptic fragments of a folded and denatured version of the same protein, the location of the unfolding is of primary interest. The same protein is being analyzed, but under different experimental conditions. In such cases, a relative deuterium level can be used rather than making a back-exchange correction to obtain the absolute number of deuterons for each exchange time-point (Fig. 1, circle 3). There are several advantages of using relative levels rather than absolute deuterium levels. First, as proper back-exchange correction relies on analysis of a totally deuterated form of the molecule of interest, a totally deuterated form must be prepared. It can be difficult to prepare such a control, especially for larger proteins. As one is never really certain that the totally deuterated control sample is really 100% deuterated at all backbone amide positions, one can never really be sure of the validity of a correction that assumes 100% deuteration. Using relative deuterium levels does not require the preparation or analysis of a totally deuterated sample. Second, because back-exchange is a complex process that depends to some extent on the sequence (Zhang & Smith, 1993), recoveries differ widely between different proteins and peptides [discussed in (Pan & Smith, 2004)]. Sequence variation is no longer a factor when using the relative method because the same sequence is being compared to itself. The only variable that has changed is the conformation of the protein(s). An added benefit of relative results is that other variables such as slight changes in buffer ph, temperature, concentration, etc. all cancel out. Ordinarily these types of variables, in addition to back-exchange variability, are significant enough to prevent the use of relative deuterium levels to compare samples obtained at different times. To take advantage of the relative method, all experiments that one wishes to compare must be completed together under identical experimental conditions. For experiments performed at the same time and under identical conditions, back-exchange is statistically the same for all B. Localizing the Label Determination of deuterium levels in whole proteins does not provide the type of local information desired. In other words, the spatial resolution is very low. To increase the resolution, proteolytic digestion is primarily used [first described by Zhang & Smith, 1993]. Because digestion of the deuterium labeled protein(s) must occur under quench conditions, acid proteases must be used. To date, the best acid-protease for these purposes is pepsin. A few complications exist however. As pepsin is a nonspecific protease (it generally cleaves at hydrophobic residues), the sites of backbone cleavage cannot be predicted from the amino acid sequence. However, pepsin will cut in the same place given the same conditions, so reproducibility is not a problem. To be certain of the identity of each peptic peptide, it becomes necessary to sequence those that are generated. Most commonly, this is accomplished using tandem MS techniques. A further complication is that peptic digestion of large proteins may produce undesirably long peptides (>15 residues long) rather than the more desirable short ones (5 10 residues). Eric Forest and colleagues recently demonstrated the use of acid proteases other than pepsin for HX MS analyses (Cravello, Lascoux, & Forest, 2003) (Fig. 1, circle 4). However, pepsin was still the most efficient protease. The use of multiple enzymes produced many overlapping fragments. Overlapping peptides are highly desirable (Garcia, Pantazatos, & Villarreal, 2004) as they increase the spatial resolution. Multiple shorter peptides were also obtained from regions where pepsin cleavage produced single, long peptide fragments. A form of pepsin that cleaves with a greater specificity would significantly improve the digestion step for HX MS. Enzyme engineering should be able to generate recombinant analogues of pepsin that have increased specificity, as seen with trypsin or other specific proteases. In addition to the complications already mentioned, another significant problem with using pepsin that is often overlooked is the interpretation of small deuterium changes in the long peptic fragments. For example, a change of 2 deuterium in a peptide of 20 amino acids cannot be attributed to changes in dynamics within the whole peptide. While curve fitting (Zhang & Smith, 1993) serves to classify the hydrogens into categories, it cannot identify them. Careful data interpretation is necessary when large fragments are involved. A method to further improve the spatial resolution is needed. C. New Fragmentation Methods It would be most desirable to be able to monitor deuterium exchange at individual amino acids with MS. Attempts have been made to use CID to fragment peptic peptides into shorter pieces (b/y ions, see Figure 4) (Deng, Pan, & Smith, 1999b; Demmers et al., 2000, 2002; Kim et al., 2001; Hoerner et al., 2004). It was originally observed that b ions from high-energy CID with argon as the collision gas yielded deuterium levels that were consistent with NMR measured deuterium levels (Deng, Pan, & Smith, 1999b; Kim et al., 2001). However, deuterium was apparently scrambled in most y ions via migration during the fragmenta- 163

7 & WALES AND ENGEN tion process. The scrambling process seemed to depend on the sequence (Demmers et al., 2002) and other multiple factors (Hoerner et al., 2004). Other work has shown that scrambling appears to be 100% in both b and y ions (Jorgensen et al., 2005). Other methods of fragmentation may offer a solution to the scrambling issue. Kaltashov and Eyles have extensively discussed the use of FTMS and electron capture dissociation (ECD) methodology towards this end (Kaltashov & Eyles, 2002a,b; Eyles & Kaltashov, 2004). It has been shown that the fragmentation of ions in ECD occurs before there is an opportunity for the energy to be randomized to a more probable bond-cleavage site (Turecek & McLafferty, 1984; Zubarev, Kelleher, & McLafferty, 1998; Horn, Ge, & McLafferty, 2000). This rapid and effective activation method may therefore result in a decrease in the hydrogen scrambling as opposed to the slower CID activation of ions. A report on the use of ECD to fragment intact proteins after HX labeling has recently appeared (Charlebois, Patrie, & Kelleher, 2003). Fragments produced by ECD of whole proteins (c/z ions, see Figure 4) may still result in incomplete sequence coverage, especially for larger proteins. It may still be advisable to digest larger proteins and protein complexes with pepsin and perform ECD on the peptides, thereby providing single amino acid resolution (Fig. 1, circle 5). Other instrumental methods have also reported on fragmentation of proteins after HX (Akashi, Naito, & Takio, 1999; Lanman et al., 2003). Unfortunately, the ECD methods described to date are limited to Fourier transform instruments for technical reasons and are therefore cost prohibitive for many research laboratories. Recent descriptions of ECD and ETD (electron transfer dissociation) in ion traps may bring this technology into the hands of researchers using standard mass analyzers (Baba et al., 2004; Syka et al., 2004). ECD/ETD technology should facilitate the eventual analysis of deuterium exchange at individual amino acids without concern for potential label scrambling. Such experiments would be a major improvement to the HX MS method (Fig. 1, circle 5). VI. WHICH KIND OF MASS SPECTROMETRY? ESI VERSUS MALDI The use of ESI-MS for the analysis of hydrogen/deuterium exchange experiments has become very popular since the mid 1990s. Approximately 80% of the articles published with respect to HX MS in the past 5 years have employed electrospray as the ionization source. ESI-MS has become the most commonly used ionization method for HX MS partially because the quenched sample is introduced directly via HPLC into the electrospray source. The advantages of using electrospray MS are described below. MALDI-MS can also be used for HX MS protein dynamics studies. E. A. Komives reported the first use of MALDI for the analysis of hydrogen exchange content in the mapping of a protein protein interface (Mandell, Falick, & Komives, 1998a). She has used MALDI HX MS to identify several binding sites in the cyclic-amp-dependent protein kinase (PKA) complex with the kinase inhibitor and ATP (Mandell, Falick, & Komives, 1998a) and to determine the antibody antigen recognition site for a monoclonal antibody raised against human thrombin (Baerga-Ortiz et al., 2002). Others have reported on the use of MALDI to probe the folding and assembly of viral capsids (Tuma et al., 2001) and to investigate protein conformational changes as a result of polyvaline and polyleucine a-helix aggregation (Hosia, Johansson, & Griffiths, 2002). Topological information about yeast F1-ATPase, a supramolecular hetero-oligomeric protein complex of 370 kda, was obtained using MALDI as the ionization method (Nazabal et al., 2003). The advantages of MALDI are that it is, in principle and generally in practice, easier than ESI for non-experienced users. All ions are singly charged and this simplifies data interpretation. There is no HPLC separation and desalting. However, the disadvantages seem to outweigh the advantages: only 20 % of all HX MS publications have used MALDI as the ionization technique. Deuterium losses are significantly higher in MALDI than in ESI, although attempts have been made to reduce these loses (Kipping & Schierhorn, 2003). While all of the peptides are present in one spectrum and no HPLC steps are required (seemingly a simplification over ESI methods), for large and complex systems, the spectra become too crowded to interpret the data. Simple HPLC separation is necessary to temporally resolve a large number of peptides. Coupling HPLC to ESI has other advantages: the HPLC step washes away deuterium label present in the amino acid side-chains (Zhang & Smith, 1993), it is compatible with all buffers and denaturants, it can afford rapid concentration of very dilute samples (although ZipTips and the like can do some concentration in MALDI analyses) and ESI can handle some buffers and matrices that would be deleterious to MALDI sample ionization. The development of new solvent-free sample preparation methods (Trimpin et al., 2001, 2002) may eliminate some of the aforementioned complications for MALDI analysis of HX samples. Eluent from a chromatographic source can be continuously deposited onto a MALDI sample stage (that has been pre-coated with matrix) by spraying the eluent at elevated temperatures (Falkenhagen et al., 2003; Falkenhagen & Weidner, 2004). The advantages of this over the conventional dried droplet method and the solvent-free grinding method are improved compatibility of analyte and matrix as well as greater sensitivity. It remains to be seen if coupling of chromatography to solventfree MALDI spotting will make the use of MALDI more attractive for HX MS analyses (Fig. 1, circle 6). VII. THINKING SMALLER Although an obvious extension of the method, the use of nano- HX MS has not yet become widespread. Off-line nano-esi-ms analyses of continuous labeled samples have been used to probe lipid interactions with transmembrane peptides (Demmers et al., 2000). The a-helical structures in the molten globule state of wild type human a-lactalbumin and several proline analogues were also investigated with off-line nano-esi-ms (Last et al., 2001). Complete online digestion, separation and ESI-MS analysis on a nano scale have also been described (Wang & Smith, 2003) and a 100-fold increase in sensitivity was reported. Nano-HX MS can be particularly valuable for proteins that are hard to obtain in large quantities or those that aggregate at higher concentrations. 164

8 PROTEIN DYNAMICS BY HYDROGEN EXCHANGE MASS SPECTROMETRY & Many rare, disease-relevant signaling proteins fit into this category. It is anticipated that more and more proteins and protein systems will require the use of nano-hx MS in the coming years (Fig. 1, circle 7). VIII. MORE COMPLEX WITH COMPLEXES IN THE MIX While analysis of proteins in vitro is providing much information about them, the Holy Grail will be to analyze proteins in vivo. Further, the analysis of protein complexes and protein machines (Gavin et al., 2002) is becoming more and more commonplace. Techniques that can work with large protein assemblies and complex matrices such as cell lysates or organelle preparations should make this dream a reality. HX MS seems positioned to make this advance and with proper method development, it may soon be possible to investigate the dynamics of a single protein from a large, multi-protein complex or a single protein in a very complex mixture like cytoplasm. In vivo hydrogen exchange has already been reported (Ghaemmaghami & Oas, 2001). Other studies investigating proteins in cell lysates have also appeared (Engen, Bradbury, & Chen, 2002). The problem in all these experiments is the separation or isolation of the protein of interest from other cellular components. When such complex systems are involved, mass alone is not sufficient to distinguish the protein of interest from the components of the matrix. Systems in which protein fragmentation occurs after initial mass separation (ECD as described above, or several tandem stages of MS and HPLC) may help alleviate this problem. Other methods to get around the complexity problem include tagging the protein of interest so that it stands out against the background of the matrix (Engen, Bradbury, & Chen, 2002). These methods, although demonstrated in principle, have yet to be used practically. It would also work to isolate the protein by affinity purification just prior to mass analysis. However, the ph requirements for HX MS hinder this possibility, although other chromatographic methods (IEX) could potentially be better suited for this purpose. HX MS methods development is moving in this direction and future investigators will no doubt solve these technical problems (Fig. 1, circle 8). IX. AUTOMATION In an ideal automated system, an operator would select a time point for analysis and automated sample preparation and analysis instrumentation would do the rest. The end result would be a graph of hydrogen exchange for a given region of a protein, also automatically represented in a 3-dimensional modeling program. While the plumbing and sample introduction (Fig. 1, circles 1, 2) parts of this ideal scenario have been automated fairly easily (Woods, 1997, 2001a,b,c), the rest has not come so easily. Data processing at the end of data acquisition is substantial and mostly has been done manually. Automation of the data analysis [mentioned in (Garcia, Pantazatos, & Villarreal, 2004)] aids in processing but is not yet ideal. Commercial versions of the software are also not available. The coming years should see more development in this area (Fig. 1, circle 9). X. RECENT EXAMPLES OF WORK USING HXMS There are many examples of analyses performed with HX MS over the past 5 years. We would like to highlight several different types of analyses with specific examples from the literature to demonstrate the range of possibilities. A. Protein Assemblies MS has opened the door to investigating the dynamics of large protein assemblies. The HIV-1 capsid assembly is one such system that has been studied recently with HX MS (Lanman et al., 2003, 2004). HX MS experiments were performed on unassembled and assembled capsid tubes to determine the putative N domain to C domain interactions in the assembled capsid tubes. Interactions between helices I and II of the N domain were identified. In addition, a previously unrecognized inter-subunit N domain C domain interaction was observed (Lanman et al., 2003). Further experiments on both the immature and mature virion revealed that helix I and helix II were involved in intersubunit interactions in both the mature and immature virion (Lanman et al., 2004). Together, these results illustrate the use of HX MS methodologies to identify possible therapeutic targets that cause disruption of viral capsid assembly. Other groups have also investigated viral capsids with HX MS (Wang, Lane, & Smith, 2001). B. Protein Dynamics Conformational changes may occur as a result of phosphorylation or by means of single amino acid mutation. Two recent studies used HX MS to describe the effects of both phosphorylation and single amino acid mutation on the COOH-terminal Src kinase (Csk), an enzyme that regulates signaling by the Srcfamily of tyrosine kinases. To provide a structural framework for understanding phosphorylation-driven protein conformational changes, HX MS was used to monitor the effects of nucleotide binding on the solution conformation of Csk in the presence of ADP and AMPPNP (a non-hydrolysable ATP analogue) (Lanman et al., 2003). The results implied that phosphorylation of Csk results in conformational changes that may influence regulatory motions in the catalytic pathway. The HX MS data also showed that the conformational states of the protein are different depending on whether substrate or product is bound (Lanman et al., 2003). To probe the conformational consequences of a single amino acid substitution at amino acid position F183 in Csk, believed to be of significant importance in the communication between the SH2 and kinase domains of Csk, HX MS experiments for three substitution analogues at this specific position were performed. Three substitutions were explored: F! G, F! Y, F!A. HX MS experiments revealed that compared to the wild-type protein, glycine substitution at F183 reduced flexibility in several peptide regions in Csk, tyrosine mutations increased flexibility, and alanine mutations showed mixed results. In addition, the data suggested that each mutant was well folded, since major regions in all 3 domains had exchange patterns that were indistinguishable from those for the wild-type protein (Lanman et al., 2004). 165
 is becoming more and more commonplace.")
9 & WALES AND ENGEN Another example of the use of HX MS to probe protein conformational dynamics involved the heat shock transcription factor s 32 (Rist et al., 2003). HX MS proved particularly valuable for this task as previous CD and fluorescence measurements were only able to detect global and local changes, respectively. As this protein tended to aggregate at high concentrations, NMR was not an available tool to study the protein solution conformation. HX MS experiments were designed to probe whether or not s 32 acted as a thermosensor. The folded states of s 32 were investigated at two separate temperatures: 37 and 428C, optimal growth conditions, and heat-stress conditions, respectively. The results indicated that there was a high degree of protein flexibility at 378C, and that there was reversible unfolding of a small structural motif at 428C. The location of this unfolded region was identified by analyzing the HX into pepsin fragments of the deuterium labeled samples. From these data, a map of the region that unfolded at elevated temperatures was produced. C. Protein Unfolding/Refolding Studies of protein folding/unfolding have traditionally been performed using the more conventional spectroscopic based techniques: NMR, IR, CD, and Fluorescence. Hydrogen exchange coupled to MS is also a useful tool for the study of protein folding/unfolding reactions. To illustrate this, some recent examples using pulse labeling HX MS are presented below. In studies of rabbit muscle triosephosphate isomerase (TIM) (Pan et al., 2004), pulse labeling denaturation experiments have revealed a bimodal isotope pattern of labeled protein in the raw MS data, which is evidence for two-state unfolding behavior. However, during the renaturation experiments there were three envelopes of isotope peaks present, suggesting that there was an intermediate in the refolding pathway. To obtain information about this intermediate, peptic digestion was performed after the pulse labeling experiment. The data showed that the intermediate was a form in which the C-terminal half was folded while the N- terminal half was not. Further, TIM folding fit a 4 þ 4 model of folding for (ba) 8 -proteins. In this set of experiments, HX MS provided location information regarding the folding intermediates as well as following both the refolding and unfolding properties. In another example, pulse labeling HX MS was employed to better characterize the molten globule intermediate state during the unfolding of the multidomain dimeric protein MM-CK (Mazon et al., 2004). In 0.8 M GdmCl, where the molten globule state of this protein was maximally populated, MM-CK exhibited a highly fluctuating structure that allowed for total deuteration. The study also used intrinsic fluorescence, ANS binding, and far- UV CD to probe the molten globule state of the protein. With HX MS, only two species were detected during GdmCl denaturation as opposed to more intermediate states detected by the conventional methods. The a-subunit of tryptophan synthase is a 29 kda, single domain protein that unfolds according to a four-state equilibrium unfolding model. There are two intermediate states, I1 and I2. The intermediate state I1 contains a significant amount of secondary structure while I2 contains no detectable secondary structure and mostly resembles the denatured state. Both of these intermediates are on-pathway kinetic species. HX MS was used to map the stable secondary structure in the I1 equilibrium intermediate (Rojsajjakul et al., 2004). Such information provided insights into the relationship between sequence, structure, and folding in the a-subunit. The identification of protected regions in the I1 intermediate was accomplished. The identified regions (most of the N-terminal region with 5 peptides regions of exception three of which are exposed loops) represented a contiguous domain (a-helices 1 3 and b-strands 1 4). It was also shown that the C-terminus, residues , was either unfolded or weakly folded in this intermediate. A refolding study of the urea denatured a-subunit of tryptophan synthase using pulse-quench HX MS has shown that there is an on pathway kinetic folding intermediate that shares a similar folded protein core with the I1 equilibrium intermediate (Wintrode et al., 2005). Together the data obtained for the equilibrium and kinetic intermediates show that the latter stages of the folding reaction for the a-subunit of tryptophan synthase are under thermodynamic control (Wintrode et al., 2005). When studying protein folding/unfolding reactions, it is not uncommon to see HX MS technologies combined with other methods. One such recent example is the integration of HX MS with a cyanylation-based methodology (Li et al., 2004) to better understand the conformation of disulfide-bonded proteins and intermediates during refolding reactions. In the cyanylationbased methodology, a protein containing disulfide bonds is trapped and the disulfide structure of a given cystinyl protein folding intermediate is identified and preserved. HX MS is then used to assess the other conformational features of the intermediate. These technologies were used to trap a 1-disulfide bond (early folding) intermediate and a 2-disulfide bond (later forming) intermediate of long Arg 3 insulin-like growth factor-i (LR 3 IGF-I) (Li et al., 2004). HX MS data showed an increasing degree of protection from exchange as a function of disulfide bond formation. There was significantly more secondary structure after formation of the second disulfide bond (specifically in helix 3). It is clear that HX MS technology is even more valuable when combined with conventional methods for the analysis of protein folding and unfolding reactions. As spectroscopic methods such as CD, IR, and Raman spectroscopy monitor globally averaged changes to protein secondary structures, and fluorescence monitors the exposure of aromatic residues to solvent, there is the possibility that they might not be sensitive to specific, local changes in protein conformation and dynamics. One can see from the above examples that HX MS can be an important complement to the traditional biophysical techniques to probe protein folding/unfolding reactions. HX MS may reveal a wealth of additional information that would otherwise go undetected. D. Binding Experiments As a result of their significance to protein folding, stability, association and function, binding interactions are under intense investigation. These interactions include protein:small molecule, protein:polypeptide, protein:lipid, protein:nucleic acid, and 166

10 PROTEIN DYNAMICS BY HYDROGEN EXCHANGE MASS SPECTROMETRY & protein:protein binding. Much information about the conformational changes that a protein undergoes during ligand binding have been determined with high-resolution X-ray crystallography or NMR structural analyses. HX MS has been used to probe the location(s) of binding sites of specific ligands on target proteins for which X-ray crystallographic and NMR methods are not applicable. Interpretation of these data, however, must be done with caution as binding is known to cause changes in protein dynamics and conformation at sites distant from the actual binding site or interface. In the discussion below there are a few examples of this observable fact [see also ref. (Mayne et al., 1992)]. An label and chase method that tries to compensate for this problem has been described (Garcia, Pantazatos, & Villarreal, 2004). There are many examples in recent years of HX MS being used in conjunction with high resolution structures to explore the organization and dynamics of complex molecular assemblies. The 42 kda eukaryotic protein actin, for example, has been investigated with HX MS (Chik & Schriemer, 2003). G-actin, F- actin (formed by polymerization of monomeric G-actin), F-actin bound to phalloidin, and a DNaseI:G-actin complex were studied to provide further information about the structure of F-actin and the structural effects of phalloidin and DNaseI ligand interactions. Note that the size and nature of polymerized actin (F-actin) excludes use of X-ray diffraction and NMR. HX MS results suggested a conformational transition from a closed to an open state of actin. The changes were localized to the phosphate binding loops upon polymerization of G-actin. Additionally, phalloidin binding to F-actin induced the monomer conformation to adopt a more G-actin-like state with the phosphate groups excluded from solution, a conformational change that inhibited phosphate release thereby reducing the rate of monomer dissociation. HX MS also provided evidence for conformational changes that occurred away from the DNaseI binding site of G-actin. These distal changes indicated a possible alteration of conformational flexibility consistent with previously published data in which the C-terminal residues were found more accessible to trypsin digestion. A study aimed at identifying regions in MAP kinases specific for binding two peptide docking motifs (DEJL and DEF) included the presentation of the distal effects of these ligand interactions (Lee et al., 2004). The data revealed that DEJL motif interactions with p38a MAPK induced enhanced backbone flexibility in the activation lip, an effect that was shown to be conserved between different MAP kinases. There were backbone conformational changes away from the region of the DEF motif interaction with ERK2 kinase. However, it remained unknown whether the interactions were attributable solely to allosteric effects or occurred as a result of alternate binding sites or nonspecific binding of the synthetic peptide to the kinase. Protein binding to small molecules is of great interest especially for development of small-molecule therapeutics. A recent HX MS study illustrating this type of experiment involved the binding of ATP to a-crystallin (Hasan, Smith, & Smith, 2002). ATP decreased the accessibility of amide hydrogens in multiple regions of both aa and ab subunits of a-crystallin. Four regions of a-crystallin, two in aa and two in ab, showed a significant decrease in the uptake of deuterium. Location information from HX MS data allowed for the comparison of the regions affected by ATP binding with proposed substrate binding sites. The authors concluded that ATP binding releases substrate by means of both direct displacement and a global conformational change. Recent HX MS binding studies between papain (target enzyme) and cystatin (thiol protease inhibitor) demonstrated that enzyme inhibitor interactions can be characterized by HX MS coupled to CID in a hexapole ion-guide using ESI-FTICR MS (Akashi & Takio, 2000). Binding of cystatin to papain reduced the flexibility throughout the papain molecule, data that are consistent with previous structural studies. HX MS protein:protein binding studies with the complex of UBC9 and SUMO-1 illustrated several key issues about protein binding experiments by HX MS (Engen, 2003). First, HXMS titrations can be used to estimate the K d for complexes. Further, backbone amide hydrogen exchange may not be altered in all proteins during complex formation, especially if the protein complex is formed primarily via electrostatic side-chain interactions. Finally, by combining site-directed mutagenesis with HX MS, much more information about the nature of complexes can be obtained. XI. LOOKING AHEAD While HX MS has come a long way in the past years, many more improvements can be made. It has only recently been widely realized that this technique can provide valuable information about proteins and protein dynamics that cannot be easily obtained with other techniques. Improvements in the method, as suggested by the circles in Figure 1, will only increase its power. We hope that many new researchers will realize this and join in the analysis of all proteins with HX MS technologies. ACKNOWLEDGMENTS We gratefully acknowledge support for this work from the National Institutes of Health (R01-GM070590, R01-GM068901, R24-CA088339, and P20-RR016480). REFERENCES Akashi S, Naito Y, Takio K Observation of hydrogen deuterium sexchange of ubiquitin by direct analysis of electrospray capillaryskimmer dissociation with fourier transform ion cyclotron resonance mass spectrometry. Anal Chem 71: Akashi S, Takio K Characterization of the interface structure of enzyme inhibitor complex by using hydrogen deuterium exchange and electrospray ionization fourier transform ion cyclotron resonance mass spectrometry. Protein Sci 9: Baba T, Hashimoto Y, Hasegawa H, Hirabayashi A, Waki I Electron capture dissociation in a radio frequency ion trap. Anal Chem 76: Baerga-Ortiz A, Hughes CA, Mandell JG, Komives EA Epitope mapping of a monoclonal antibody against human thrombin by h/dexchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein. Protein Sci 11:

11 & WALES AND ENGEN Bai Y, Milne JS, Mayne L, Englander SW Primary structure effects on peptide group hydrogen exchange. Proteins Struct Funct Genet 17: Bai YW, Milne JS, Mayne L, Englander SW Protein stability parameters measured by hydrogen exchange. Proteins Struct Funct Genet 20:4 14. Bai Y, Sosnick TR, Mayne L, Englander SW Protein folding intermediates: Native-state hydrogen exchange. Science 269: Chamberlain AK, Marqusee S Molten globule unfolding monitored by hydrogen exchange in urea. Biochemistry 37: Charlebois JP, Patrie SM, Kelleher NL Electron capture dissociation and 13c,15n depletion for deuterium localization in intact proteins after solution-phase exchange. Anal Chem 75: Chen J, Walter S, Horwich AL, Smith DL Folding of malate dehydrogenase inside the groel-groes cavity. Nat Struct Biol 8: Chik JK, Schriemer DC Hydrogen/deuterium exchange mass spectrometry of actin in various biochemical contexts. J Mol Biol 334: Chowdhury SK, Katta V, Chait BT Probing conformational changes in proteins by mass spectrometry. J Am Chem Soc 112: Clarke J, Itzhaki LS, Fersht AR Hydrogen exchange at equilibrium: A short cut for analysing protein-folding pathways? TIBS 22: Cravello L, Lascoux D, Forest E Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun Mass Spectrom 17: Demmers JA, Haverkamp J, Heck AJ, Koeppe RE, 2nd, Killian JA Electrospray ionization mass spectrometry as a tool to analyze hydrogen/deuterium exchange kinetics of transmembrane peptides in lipid bilayers. Proc Natl Acad Sci USA 97: Demmers JA, Rijkers DT, Haverkamp J, Killian JA, Heck AJ Factors affecting gas-phase deuterium scrambling in peptide ions and their implications for protein structure determination. J Am Chem Soc 124: Deng Y, Pan H, Smith DL. 1999a. Comparison of continuous and pulsed labeling amide hydrogen exchange/mass spectrometry for studies of protein dynamics. J Am Soc Mass Spectrom 10: Deng Y, Pan H, Smith DL. 1999b. Selective isotope labeling demonstrates that hydrogen exchange at individual peptide amide linkages can be determined by collision-induced dissocation mass spectrometry. J Am Chem Soc 121: Deng Y, Smith DL Identification of unfolding domains in large proteins by their unfolding rates. Biochemistry 37: Deng Y, Smith DL Rate and equilibrium constants for protein unfolding and refolding determined by hydrogen exchange-mass spectrometry. Anal Biochem 276: Dyson HJ, Wright PE Unfolded proteins and protein folding studied by NMR. Chem Rev 104: Engen JR Analysis of protein complexes with hydrogen exchange and mass spectrometry. Analyst (London) 128: Engen JR, Bradbury EM, Chen X Using stable-isotope-labeled proteins for hydrogen exchange studies in complex mixtures. Anal Chem 74: Engen JR, Smith DL Investigating the higher order structure of proteins: Hydrogen exchange, proteolytic fragmentation & mass spectrometry. Methods in Mol Biol 146: Engen JR, Smith DL Investigating protein structure and dynamics by hydrogen exchange ms. Anal Chem 73:256A 265A. Engen JR, Smithgall TE, Gmeiner WH, Smith DL Identification and localization of slow, natural, cooperative unfolding in the hematopoietic cell kinase sh3 domain by amide hydrogen exchange and mass spectrometry. Biochemistry 36: Englander SW, Kallenbach NR Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys 16: Eyles SJ, Kaltashov IA Methods to study protein dynamics and folding by mass spectrometry. Methods 34: Falkenhagen J, Weidner SM nd ASMS Conference on Mass Spectrometry and Allied Topics, Nashville, Tennessee, May 23 27, Falkenhagen J, Jancke H, Kruger RP, Rikowski E, Schulz G Characterization of silsesquioxanes by size-exclusion chromatography and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 17: Garcia RA, Pantazatos D, Villarreal FJ Hydrogen/deuterium exchange mass spectrometry for investigating protein ligand interactions. Assay Drug Dev Technol 2: Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti- Furga G Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: Ghaemmaghami S, Oas TG Quantitative protein stability measurement in vivo. Nat Struct Biol 8: Hasan A, Smith DL, Smith JB Alpha-crystallin regions affected by adenosine 5 0 -triphosphate identified by hydrogen deuterium exchange. Biochemistry 41: Hoerner JK, Xiao H, Dobo A, Kaltashov IA Is there hydrogen scrambling in the gas phase? Energetic and structural determinants of proton mobility within protein ions. J Am Chem Soc 126: Hoofnagle AN, Resing KA, Ahn NG Protein analysis by hydrogen exchange mass spectrometry. Annu Rev Biophys Biomol Struct 32: Hoofnagle AN, Resing KA, Ahn NG Practical methods for deuterium exchange/mass spectrometry. Methods Mol Biol 250: Horn DM, Ge Y, McLafferty FW Activated ion electron capture dissociation for mass spectral sequencing of larger (42 kda) proteins. Anal Chem 72: Hosia W, Johansson J, Griffiths WJ Hydrogen/deuterium exchange and aggregation of a polyvaline and a polyleucine alpha-helix investigated by matrix-assisted laser desorption ionization mass spectrometry. Mol Cell Proteomics 1: Hvidt A, Nielsen SO Hydrogen exchange in proteins. Adv Protein Chem 21: Itzhaki LS, Neira JL, Fersht AR Hydrogen exchange in chymotrypsin inhibitor 2 probed by denaturants and temperature. J Mol Biol 270: Jorgensen TJ, Gardsvoll H, Ploug M, Roepstorff P Intramolecular migration of amide hydrogens in protonated peptides upon collisional activation. J Am Chem Soc 127: Kaltashov IA, Eyles SJ. 2002a. Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spectrom Rev 21: Kaltashov IA, Eyles SJ. 2002b. Crossing the phase boundary to study protein dynamics and function: Combination of amide hydrogen exchange in solution and ion fragmentation in the gas phase. J Mass Spectrom 37: Katta V, Chait BT Conformational changes in proteins probed by hydrogen-exchange electrospray-ionization mass spectrometry. Rapid Commun Mass Spectrom 5: Kim K-S, Woodward C Protein internal flexibility and global stability: Effect of urea on hydrogen exchange rates of bovine pancreatic trypsin inhibitor. Biochemistry 32:

12 PROTEIN DYNAMICS BY HYDROGEN EXCHANGE MASS SPECTROMETRY & Kim MY, Maier CS, Reed DJ, Deinzer ML Site-specific amide hydrogen/deuterium exchange in E. coli thioredoxins measured by electrospray ionization mass spectrometry. J Am Chem Soc 123: Kipping M, Schierhorn A Improving hydrogen/deuterium exchange mass spectrometry by reduction of the back-exchange effect. J Mass Spectrom 38: Konerman L, Simmons DA Protein-folding kinetics and mechanisms studied by pulsed-labeling and mass spectrometry. Mass Spectrom Rev 22:1 26. Krishna MM, Hoang L, Lin Y, Englander SW Hydrogen exchange methods to study protein folding. Methods 34: Lanman J, Lam TT, Barnes S, Sakalian M, Emmett MR, Marshall AG, Prevelige PE, Jr Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J Mol Biol 325: Lanman J, Lam TT, Emmett MR, Marshall AG, Sakalian M, Prevelige PE, Jr Key interactions in HIV-1 maturation identified by hydrogen deuterium exchange. Nat Struct Mol Biol 11: Last AM, Schulman BA, Robinson CV, Redfield C Probing subtle differences in the hydrogen exchange behavior of variants of the human alpha-lactalbumin molten globule using mass spectrometry. J Mol Biol 311: Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG Docking motif interactions in map kinases revealed by hydrogen exchange mass spectrometry. Mol Cell 14: Li X, Chou YT, Husain R, Watson JT Integration of hydrogen/ deuterium exchange and cyanylation-based methodology for conformational studies of cystinyl proteins. Anal Biochem 331: Loh SN, Rohl CA, Kiefhaber T, Baldwin RL A general two-process model describes the hydrogen exchange behavior of RNAse a in unfolding conditions. Proc Natl Acad Sci USA 93: Mandell JG, Falick AM, Komives EA. 1998a. Identification of protein protein interfaces by decreased amide proton solvent accessibility. Proc Natl Acad Sci USA 95: Mandell JG, Falick AM, Komives EA. 1998b. Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal Chem 70: Mayne L, Paterson Y, Cerasoli D, Englander SW Effect of antibody binding on protein motions studied by hydrogen-exchange labeling and two-dimensional NMR. Biochemistry 31: Mayo SL, Baldwin RL Guanidinium chloride induction of partial unfolding in amide proton exchange in RNAse a. Science 262: Mazon H, Marcillat O, Forest E, Smith DL, Vial C Conformational dynamics of the gdmhcl-induced molten globule state of creatine kinase monitored by hydrogen exchange and mass spectrometry. Biochemistry 43: Miller DW, Dill KA A statistical mechanical model for hydrogen exchange in globular proteins. Protein Sci 4: Miranker A, Robinson CV, Radford SE, Aplin RT, Dobson CM Detection of transient protein folding populations by mass spectrometry. Science 262: Nazabal A, Laguerre M, Schmitter JM, Vaillier J, Chaignepain S, Velours J Hydrogen/deuterium exchange on yeast atpase supramolecular protein complex analyzed at high sensitivity by MALDI mass spectrometry. J Am Soc Mass Spectrom 14: Nishimura C, Wright PE, Dyson HJ Role of the b helix in early folding events in apomyoglobin: Evidence from site-directed mutagenesis for native-like long range interactions. J Mol Biol 334: Pan H, Raza AS, Smith DL Equilibrium and kinetic folding of rabbit muscle triosephosphate isomerase by hydrogen exchange mass spectrometry. J Mol Biol 336: Pan H, Smith DL Amide hydrogen exchange/mass spectrometry applied to cooperative protein folding: Equilibrium unfolding of Staphylococcus aureus aldolase. Methods Enzymol 380: Resing KA, Hoofnagle AN, Ahn NG Modeling deuterium exchange behavior of erk2 using pepsin mapping to probe secondary structure. J Am Soc Mass Spectrom 10: Rist W, Jorgensen TJ, Roepstorff P, Bukau B, Mayer MP Mapping temperature-induced conformational changes in the Escherichia coli heat shock transcription factor sigma 32 by amide hydrogen exchange. J Biol Chem 278: Roepstorff P, Fohlman J Proposal for common nomenclature for sequence ions in mass spectra of peptides. Biomed Mass Spectrom 11:601. Rojsajjakul T, Wintrode P, Vadrevu R, Robert Matthews C, Smith DL Multi-state unfolding of the alpha subunit of tryptophan synthase, a tim barrel protein: Insights into the secondary structure of the stable equilibrium intermediates by hydrogen exchange mass spectrometry. J Mol Biol 341: Smith DL, Deng Y, Zhang Z Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J Mass Spectrom 32: Swint-Kruse L, Robertson AD Temperature and ph dependence of hydrogen exchange and global stability for ovomucoid third domain. Biochemistry 35: Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA 101: Trimpin S, Rouhanipour A, Az R, Rader HJ, Mullen K New aspects in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry: A universal solvent-free sample preparation. Rapid Commun Mass Spectrom 15: Trimpin S, Grimsdale AC, Rader HJ, Mullen K Characterization of an insoluble poly(9,9-diphenyl-2,7-fluorene) by solvent-free sample preparation for MALDI-TOF mass spectrometry. Anal Chem 74: Tuma R, Coward LU, Kirk MC, Barnes S, Prevelige PE, Jr Hydrogen deuterium exchange as a probe of folding and assembly in viral capsids. J Mol Biol 306: Turecek F, McLafferty FW Non-ergodic behavior in acetone enol ion dissociations. J Am Chem Soc 106: Wang L, Lane LC, Smith DL Detecting structural changes in viral capsids by hydrogen exchange and mass spectrometry. Protein Sci 10: Wang L, Smith DL Downsizing improves sensitivity 100-fold for hydrogen exchange-mass spectrometry. Anal Biochem 314: Wintrode PL, Friedrich KL, Vierling E, Smith JB, Smith DL Solution structure and dynamics of a heat shock protein assembly probed by hydrogen exchange and mass spectrometry. Biochemistry 42: Wintrode PL, Rojsajjakul T, Vadrevu R, Matthews CR, Smith DL An obligatory intermediate controls the folding of the alpha-subunit of tryptophan synthase, a TIM barrel protein. J Mol Biol 347: Woods VL, Jr Method for characterization of the fine structure of protein binding sites. In. USA: The Regents of the University of California. US patent #5,658,739. Woods VL, Jr. 2001a. Methods for the high-resolution identification of solvent-accessible amide hydrogens in polypeptides or proteins and for characterization of the fine structure of protein binding sites. In. USA: Carta Proteomics, Inc. US patent 6,291,

13 & WALES AND ENGEN Woods VL, Jr. 2001b. Methods for identifying hot-spot residues of binding proteins and small compounds that bind to the same. In. USA: Carta Proteomics, Inc. US patent 6,599,707. Woods VL, Jr. 2001c. Method for characterization of the fine structure of protein binding sites. In. USA: Carta Proteomics, Inc. US patent 6,331,400. Woodward C, Simon I, Tüchsen E Hydrogen exchange and the dynamic structure of proteins. Mol Cell Biochem 48: Wu Y, Engen JR What mass spectrometry can reveal about protein function. Analyst 129: Yang H, Smith DL Kinetics of cytochrome c folding examined by hydrogen exchange and mass spectrometry. Biochemistry 36: Zhang Z, Smith DL Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci 2: Zubarev RA, Kelleher NL, McLafferty FW Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc 120: Dr. Thomas E. Wales received his B.A. degree in Chemistry and Spanish (1998) from Assumption College and his Ph.D. in Analytical Chemistry (2003) from Duke University. He is currently a postdoctoral fellow with John R. Engen at the University of New Mexico. His research has focused on the analysis of the protein backbone from both a chemical and structural perspective. Currently he is investigating the minute time-scale unfolding of protein backbone motions using hydrogen-deuterium exchange mass spectrometry. Dr. John R. Engen received B.S. degrees in Molecular Biology (1994) and Biochemistry (1995) from Union College and his Ph.D. in Analytical Chemistry (1999) from the University of Nebraska-Lincoln. As an EMBO Fellow, he did postdoctoral work (2000) in molecular biology and cellular signaling at the European Molecular Biology Laboratory in Heidelberg, Germany followed by a second postdoctoral appointment (2001) at Los Alamos National Laboratory. He was appointed an Assistant Professor of Chemistry at the University of New Mexico in January His research centers around the analysis of protein function with mass spectrometry, specifically hydrogen exchange analysis of structural activation in oncogenic kinases and protein folding during cellular processes. 170

Application of a New Immobilization H/D Exchange Protocol: A Calmodulin Study
 Application of a New Immobilization H/D Exchange Protocol: A Calmodulin Study Jiang Zhao; Mei Zhu; Daryl E. Gilblin; Michael L. Gross Washington University Center for Biomrdical and Bioorganic Mass Spectrometry:
Application of a New Immobilization H/D Exchange Protocol: A Calmodulin Study Jiang Zhao; Mei Zhu; Daryl E. Gilblin; Michael L. Gross Washington University Center for Biomrdical and Bioorganic Mass Spectrometry:
Analysis of structural dynamics by H/D-exchange coupled to mass spectrometry HDX-MS
 Analysis of structural dynamics by H/D-exchange coupled to mass spectrometry () New Approaches in Drug Design & Discovery 2014 25 th of March 2014 Introduction What are the challenges in structure-based
Analysis of structural dynamics by H/D-exchange coupled to mass spectrometry () New Approaches in Drug Design & Discovery 2014 25 th of March 2014 Introduction What are the challenges in structure-based
Chapter 3. Protein Structure and Function
 Chapter 3 Protein Structure and Function Broad functional classes So Proteins have structure and function... Fine! -Why do we care to know more???? Understanding functional architechture gives us POWER
Chapter 3 Protein Structure and Function Broad functional classes So Proteins have structure and function... Fine! -Why do we care to know more???? Understanding functional architechture gives us POWER
(c) How would your answers to problem (a) change if the molecular weight of the protein was 100,000 Dalton?
 How would your answers to problem (a) change if the molecular weight of the protein was 100,000 Dalton?") Problem 1. (12 points total, 4 points each) The molecular weight of an unspecified protein, at physiological conditions, is 70,000 Dalton, as determined by sedimentation equilibrium measurements and by
Problem 1. (12 points total, 4 points each) The molecular weight of an unspecified protein, at physiological conditions, is 70,000 Dalton, as determined by sedimentation equilibrium measurements and by
Methods for Protein Analysis
 Methods for Protein Analysis 1. Protein Separation Methods The following is a quick review of some common methods used for protein separation: SDS-PAGE (SDS-polyacrylamide gel electrophoresis) separates
Methods for Protein Analysis 1. Protein Separation Methods The following is a quick review of some common methods used for protein separation: SDS-PAGE (SDS-polyacrylamide gel electrophoresis) separates
Mass Spectrometry Signal Calibration for Protein Quantitation
 Cambridge Isotope Laboratories, Inc. www.isotope.com Proteomics Mass Spectrometry Signal Calibration for Protein Quantitation Michael J. MacCoss, PhD Associate Professor of Genome Sciences University of
Cambridge Isotope Laboratories, Inc. www.isotope.com Proteomics Mass Spectrometry Signal Calibration for Protein Quantitation Michael J. MacCoss, PhD Associate Professor of Genome Sciences University of
8/20/2012 H C OH H R. Proteins
 Proteins Rubisco monomer = amino acids 20 different amino acids polymer = polypeptide protein can be one or more polypeptide chains folded & bonded together large & complex 3-D shape hemoglobin Amino acids
Proteins Rubisco monomer = amino acids 20 different amino acids polymer = polypeptide protein can be one or more polypeptide chains folded & bonded together large & complex 3-D shape hemoglobin Amino acids
Supplementary Materials for
 www.sciencesignaling.org/cgi/content/full/7/339/ra80/dc1 Supplementary Materials for Manipulation of receptor oligomerization as a strategy to inhibit signaling by TNF superfamily members Julia T. Warren,
www.sciencesignaling.org/cgi/content/full/7/339/ra80/dc1 Supplementary Materials for Manipulation of receptor oligomerization as a strategy to inhibit signaling by TNF superfamily members Julia T. Warren,
Review of Chemical Equilibrium 7.51 September 1999. free [A] (µm)
![Review of Chemical Equilibrium 7.51 September 1999. free [A] (µm)](/thumbs/40/21435474.jpg "Review of Chemical Equilibrium 7.51 September 1999. free [A] (µm)") Review of Chemical Equilibrium 7.51 September 1999 Equilibrium experiments study how the concentration of reaction products change as a function of reactant concentrations and/or reaction conditions. For
Review of Chemical Equilibrium 7.51 September 1999 Equilibrium experiments study how the concentration of reaction products change as a function of reactant concentrations and/or reaction conditions. For
http://faculty.sau.edu.sa/h.alshehri
 http://faculty.sau.edu.sa/h.alshehri Definition: Proteins are macromolecules with a backbone formed by polymerization of amino acids. Proteins carry out a number of functions in living organisms: - They
http://faculty.sau.edu.sa/h.alshehri Definition: Proteins are macromolecules with a backbone formed by polymerization of amino acids. Proteins carry out a number of functions in living organisms: - They
Papers listed: Cell2. This weeks papers. Chapt 4. Protein structure and function
 Papers listed: Cell2 During the semester I will speak of information from several papers. For many of them you will not be required to read these papers, however, you can do so for the fun of it (and it
Papers listed: Cell2 During the semester I will speak of information from several papers. For many of them you will not be required to read these papers, however, you can do so for the fun of it (and it
Peptide bonds: resonance structure. Properties of proteins: Peptide bonds and side chains. Dihedral angles. Peptide bond. Protein physics, Lecture 5
 Protein physics, Lecture 5 Peptide bonds: resonance structure Properties of proteins: Peptide bonds and side chains Proteins are linear polymers However, the peptide binds and side chains restrict conformational
Protein physics, Lecture 5 Peptide bonds: resonance structure Properties of proteins: Peptide bonds and side chains Proteins are linear polymers However, the peptide binds and side chains restrict conformational
Built from 20 kinds of amino acids
 Built from 20 kinds of amino acids Each Protein has a three dimensional structure. Majority of proteins are compact. Highly convoluted molecules. Proteins are folded polypeptides. There are four levels
Built from 20 kinds of amino acids Each Protein has a three dimensional structure. Majority of proteins are compact. Highly convoluted molecules. Proteins are folded polypeptides. There are four levels
18.2 Protein Structure and Function: An Overview
 18.2 Protein Structure and Function: An Overview Protein: A large biological molecule made of many amino acids linked together through peptide bonds. Alpha-amino acid: Compound with an amino group bonded
18.2 Protein Structure and Function: An Overview Protein: A large biological molecule made of many amino acids linked together through peptide bonds. Alpha-amino acid: Compound with an amino group bonded
Biochemistry - I. Prof. S. Dasgupta Department of Chemistry Indian Institute of Technology, Kharagpur Lecture-11 Enzyme Mechanisms II
 Biochemistry - I Prof. S. Dasgupta Department of Chemistry Indian Institute of Technology, Kharagpur Lecture-11 Enzyme Mechanisms II In the last class we studied the enzyme mechanisms of ribonuclease A
Biochemistry - I Prof. S. Dasgupta Department of Chemistry Indian Institute of Technology, Kharagpur Lecture-11 Enzyme Mechanisms II In the last class we studied the enzyme mechanisms of ribonuclease A
Proteomics in Practice
 Reiner Westermeier, Torn Naven Hans-Rudolf Höpker Proteomics in Practice A Guide to Successful Experimental Design 2008 Wiley-VCH Verlag- Weinheim 978-3-527-31941-1 Preface Foreword XI XIII Abbreviations,
Reiner Westermeier, Torn Naven Hans-Rudolf Höpker Proteomics in Practice A Guide to Successful Experimental Design 2008 Wiley-VCH Verlag- Weinheim 978-3-527-31941-1 Preface Foreword XI XIII Abbreviations,
A. 'Hypersensitive' peptide bonds and autodegradation of proteins
 ABSTRACT A. 'Hypersensitive' peptide bonds and autodegradation of proteins Several pure proteins, which gave a single band on electrophoretic analysis, when stored for a long time, were found to be partially
ABSTRACT A. 'Hypersensitive' peptide bonds and autodegradation of proteins Several pure proteins, which gave a single band on electrophoretic analysis, when stored for a long time, were found to be partially
The Organic Chemistry of Amino Acids, Peptides, and Proteins
 Essential rganic Chemistry Chapter 16 The rganic Chemistry of Amino Acids, Peptides, and Proteins Amino Acids a-amino carboxylic acids. The building blocks from which proteins are made. H 2 N C 2 H Note:
Essential rganic Chemistry Chapter 16 The rganic Chemistry of Amino Acids, Peptides, and Proteins Amino Acids a-amino carboxylic acids. The building blocks from which proteins are made. H 2 N C 2 H Note:
--not necessarily a protein! (all proteins are polypeptides, but the converse is not true)
") 00Note Set 5b 1 PEPTIDE BONDS AND POLYPEPTIDES OLIGOPEPTIDE: --chain containing only a few amino acids (see tetrapaptide, Fig 5.9) POLYPEPTIDE CHAINS: --many amino acids joined together --not necessarily
00Note Set 5b 1 PEPTIDE BONDS AND POLYPEPTIDES OLIGOPEPTIDE: --chain containing only a few amino acids (see tetrapaptide, Fig 5.9) POLYPEPTIDE CHAINS: --many amino acids joined together --not necessarily
FTIR Analysis of Protein Structure
 FTIR Analysis of Protein Structure Warren Gallagher A. Introduction to protein structure The first structures of proteins at an atomic resolution were determined in the late 1950 s. 1 From that time to
FTIR Analysis of Protein Structure Warren Gallagher A. Introduction to protein structure The first structures of proteins at an atomic resolution were determined in the late 1950 s. 1 From that time to
Chapter 8: Energy and Metabolism
 Chapter 8: Energy and Metabolism 1. Discuss energy conversions and the 1 st and 2 nd law of thermodynamics. Be sure to use the terms work, potential energy, kinetic energy, and entropy. 2. What are Joules
Chapter 8: Energy and Metabolism 1. Discuss energy conversions and the 1 st and 2 nd law of thermodynamics. Be sure to use the terms work, potential energy, kinetic energy, and entropy. 2. What are Joules
NO CALCULATORS OR CELL PHONES ALLOWED
 Biol 205 Exam 1 TEST FORM A Spring 2008 NAME Fill out both sides of the Scantron Sheet. On Side 2 be sure to indicate that you have TEST FORM A The answers to Part I should be placed on the SCANTRON SHEET.
Biol 205 Exam 1 TEST FORM A Spring 2008 NAME Fill out both sides of the Scantron Sheet. On Side 2 be sure to indicate that you have TEST FORM A The answers to Part I should be placed on the SCANTRON SHEET.
Protein Physics. A. V. Finkelstein & O. B. Ptitsyn LECTURE 1
 Protein Physics A. V. Finkelstein & O. B. Ptitsyn LECTURE 1 PROTEINS Functions in a Cell MOLECULAR MACHINES BUILDING BLOCKS of a CELL ARMS of a CELL ENZYMES - enzymatic catalysis of biochemical reactions
Protein Physics A. V. Finkelstein & O. B. Ptitsyn LECTURE 1 PROTEINS Functions in a Cell MOLECULAR MACHINES BUILDING BLOCKS of a CELL ARMS of a CELL ENZYMES - enzymatic catalysis of biochemical reactions
Lecture 11 Enzymes: Kinetics
 Lecture 11 Enzymes: Kinetics Reading: Berg, Tymoczko & Stryer, 6th ed., Chapter 8, pp. 216-225 Key Concepts Kinetics is the study of reaction rates (velocities). Study of enzyme kinetics is useful for
Lecture 11 Enzymes: Kinetics Reading: Berg, Tymoczko & Stryer, 6th ed., Chapter 8, pp. 216-225 Key Concepts Kinetics is the study of reaction rates (velocities). Study of enzyme kinetics is useful for
Energy & Enzymes. Life requires energy for maintenance of order, growth, and reproduction. The energy living things use is chemical energy.
 Energy & Enzymes Life requires energy for maintenance of order, growth, and reproduction. The energy living things use is chemical energy. 1 Energy exists in two forms - potential and kinetic. Potential
Energy & Enzymes Life requires energy for maintenance of order, growth, and reproduction. The energy living things use is chemical energy. 1 Energy exists in two forms - potential and kinetic. Potential
PROTEIN SEQUENCING. First Sequence
 PROTEIN SEQUENCING First Sequence The first protein sequencing was achieved by Frederic Sanger in 1953. He determined the amino acid sequence of bovine insulin Sanger was awarded the Nobel Prize in 1958
PROTEIN SEQUENCING First Sequence The first protein sequencing was achieved by Frederic Sanger in 1953. He determined the amino acid sequence of bovine insulin Sanger was awarded the Nobel Prize in 1958
AP BIOLOGY 2008 SCORING GUIDELINES
 AP BIOLOGY 2008 SCORING GUIDELINES Question 1 1. The physical structure of a protein often reflects and affects its function. (a) Describe THREE types of chemical bonds/interactions found in proteins.
AP BIOLOGY 2008 SCORING GUIDELINES Question 1 1. The physical structure of a protein often reflects and affects its function. (a) Describe THREE types of chemical bonds/interactions found in proteins.
Lecture 13-14 Conformation of proteins Conformation of a protein three-dimensional structure native state. native condition
 Lecture 13-14 Conformation of proteins Conformation of a protein refers to the three-dimensional structure in its native state. There are many different possible conformations for a molecule as large as
Lecture 13-14 Conformation of proteins Conformation of a protein refers to the three-dimensional structure in its native state. There are many different possible conformations for a molecule as large as
A. A peptide with 12 amino acids has the following amino acid composition: 2 Met, 1 Tyr, 1 Trp, 2 Glu, 1 Lys, 1 Arg, 1 Thr, 1 Asn, 1 Ile, 1 Cys
 Questions- Proteins & Enzymes A. A peptide with 12 amino acids has the following amino acid composition: 2 Met, 1 Tyr, 1 Trp, 2 Glu, 1 Lys, 1 Arg, 1 Thr, 1 Asn, 1 Ile, 1 Cys Reaction of the intact peptide
Questions- Proteins & Enzymes A. A peptide with 12 amino acids has the following amino acid composition: 2 Met, 1 Tyr, 1 Trp, 2 Glu, 1 Lys, 1 Arg, 1 Thr, 1 Asn, 1 Ile, 1 Cys Reaction of the intact peptide
Laboration 1. Identifiering av proteiner med Mass Spektrometri. Klinisk Kemisk Diagnostik
 Laboration 1 Identifiering av proteiner med Mass Spektrometri Klinisk Kemisk Diagnostik Sven Kjellström 2014 kjellstrom.sven@gmail.com 0702-935060 Laboration 1 Klinisk Kemisk Diagnostik Identifiering av
Laboration 1 Identifiering av proteiner med Mass Spektrometri Klinisk Kemisk Diagnostik Sven Kjellström 2014 kjellstrom.sven@gmail.com 0702-935060 Laboration 1 Klinisk Kemisk Diagnostik Identifiering av
IV. -Amino Acids: carboxyl and amino groups bonded to -Carbon. V. Polypeptides and Proteins
 IV. -Amino Acids: carboxyl and amino groups bonded to -Carbon A. Acid/Base properties 1. carboxyl group is proton donor! weak acid 2. amino group is proton acceptor! weak base 3. At physiological ph: H
IV. -Amino Acids: carboxyl and amino groups bonded to -Carbon A. Acid/Base properties 1. carboxyl group is proton donor! weak acid 2. amino group is proton acceptor! weak base 3. At physiological ph: H
Introduction to Proteomics 1.0
 Introduction to Proteomics 1.0 CMSP Workshop Tim Griffin Associate Professor, BMBB Faculty Director, CMSP Objectives Why are we here? For participants: Learn basics of MS-based proteomics Learn what s
Introduction to Proteomics 1.0 CMSP Workshop Tim Griffin Associate Professor, BMBB Faculty Director, CMSP Objectives Why are we here? For participants: Learn basics of MS-based proteomics Learn what s
CSC 2427: Algorithms for Molecular Biology Spring 2006. Lecture 16 March 10
 CSC 2427: Algorithms for Molecular Biology Spring 2006 Lecture 16 March 10 Lecturer: Michael Brudno Scribe: Jim Huang 16.1 Overview of proteins Proteins are long chains of amino acids (AA) which are produced
CSC 2427: Algorithms for Molecular Biology Spring 2006 Lecture 16 March 10 Lecturer: Michael Brudno Scribe: Jim Huang 16.1 Overview of proteins Proteins are long chains of amino acids (AA) which are produced
Thermo Scientific PepFinder Software A New Paradigm for Peptide Mapping
 Thermo Scientific PepFinder Software A New Paradigm for Peptide Mapping For Conclusive Characterization of Biologics Deep Protein Characterization Is Crucial Pharmaceuticals have historically been small
Thermo Scientific PepFinder Software A New Paradigm for Peptide Mapping For Conclusive Characterization of Biologics Deep Protein Characterization Is Crucial Pharmaceuticals have historically been small
Copyright The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Two Forms of Energy
 Module 2D - Energy and Metabolism Objective # 19 All living organisms require energy for survival. In this module we will examine some general principles about chemical reactions and energy usage within
Module 2D - Energy and Metabolism Objective # 19 All living organisms require energy for survival. In this module we will examine some general principles about chemical reactions and energy usage within
Hydrogen Bonds The electrostatic nature of hydrogen bonds
 Hydrogen Bonds Hydrogen bonds have played an incredibly important role in the history of structural biology. Both the structure of DNA and of protein a-helices and b-sheets were predicted based largely
Hydrogen Bonds Hydrogen bonds have played an incredibly important role in the history of structural biology. Both the structure of DNA and of protein a-helices and b-sheets were predicted based largely
Chemical Exchange in NMR Spectroscopy
 COURSE#1022: Biochemical Applications of NMR Spectroscopy http://www.bioc.aecom.yu.edu/labs/girvlab/nmr/course/ Chemical Exchange in NMR Spectroscopy LAST UPDATE: 3/28/2012 1 References Bain, A. D. (2003).
COURSE#1022: Biochemical Applications of NMR Spectroscopy http://www.bioc.aecom.yu.edu/labs/girvlab/nmr/course/ Chemical Exchange in NMR Spectroscopy LAST UPDATE: 3/28/2012 1 References Bain, A. D. (2003).
Amino Acids and Proteins
 Amino Acids and Proteins Proteins are composed of amino acids. There are 20 amino acids commonly found in proteins. All have: N2 C α R COO Amino acids at neutral p are dipolar ions (zwitterions) because
Amino Acids and Proteins Proteins are composed of amino acids. There are 20 amino acids commonly found in proteins. All have: N2 C α R COO Amino acids at neutral p are dipolar ions (zwitterions) because
Chapter 8: An Introduction to Metabolism
 Chapter 8: An Introduction to Metabolism Name Period Concept 8.1 An organism s metabolism transforms matter and energy, subject to the laws of thermodynamics 1. Define metabolism. The totality of an organism
Chapter 8: An Introduction to Metabolism Name Period Concept 8.1 An organism s metabolism transforms matter and energy, subject to the laws of thermodynamics 1. Define metabolism. The totality of an organism
Disulfide Bonds at the Hair Salon
 Disulfide Bonds at the Hair Salon Three Alpha Helices Stabilized By Disulfide Bonds! In order for hair to grow 6 inches in one year, 9 1/2 turns of α helix must be produced every second!!! In some proteins,
Disulfide Bonds at the Hair Salon Three Alpha Helices Stabilized By Disulfide Bonds! In order for hair to grow 6 inches in one year, 9 1/2 turns of α helix must be produced every second!!! In some proteins,
Helices From Readily in Biological Structures
 The α Helix and the β Sheet Are Common Folding Patterns Although the overall conformation each protein is unique, there are only two different folding patterns are present in all proteins, which are α
The α Helix and the β Sheet Are Common Folding Patterns Although the overall conformation each protein is unique, there are only two different folding patterns are present in all proteins, which are α
Protein Dynamics Intro
 Protein Dynamics Intro From rigid structures to motions on energy landscapes Do you all remember Anfinsen? What concept now associated with his name made Anfinsen famous? Right, it is the concept that
Protein Dynamics Intro From rigid structures to motions on energy landscapes Do you all remember Anfinsen? What concept now associated with his name made Anfinsen famous? Right, it is the concept that
Advanced Medicinal & Pharmaceutical Chemistry CHEM 5412 Dept. of Chemistry, TAMUK
 Advanced Medicinal & Pharmaceutical Chemistry CHEM 5412 Dept. of Chemistry, TAMUK Dai Lu, Ph.D. dlu@tamhsc.edu Tel: 361-221-0745 Office: RCOP, Room 307 Drug Discovery and Development Drug Molecules Medicinal
Advanced Medicinal & Pharmaceutical Chemistry CHEM 5412 Dept. of Chemistry, TAMUK Dai Lu, Ph.D. dlu@tamhsc.edu Tel: 361-221-0745 Office: RCOP, Room 307 Drug Discovery and Development Drug Molecules Medicinal
VTT TECHNICAL RESEARCH CENTRE OF FINLAND
 Figure from: http://www.embl.de/nmr/sattler/teaching Why NMR (instead of X ray crystallography) a great number of macromolecules won't crystallize) natural environmant (water) ligand binding and inter
Figure from: http://www.embl.de/nmr/sattler/teaching Why NMR (instead of X ray crystallography) a great number of macromolecules won't crystallize) natural environmant (water) ligand binding and inter
Overview'of'Solid-Phase'Peptide'Synthesis'(SPPS)'and'Secondary'Structure'Determination'by'FTIR'
'and'Secondary'Structure'Determination'by'FTIR'") verviewofsolid-phasepeptidesynthesis(spps)andsecondarystructuredeterminationbyftir Introduction Proteinsareubiquitousinlivingorganismsandcells,andcanserveavarietyoffunctions.Proteinscanactas enzymes,hormones,antibiotics,receptors,orserveasstructuralsupportsintissuessuchasmuscle,hair,and
verviewofsolid-phasepeptidesynthesis(spps)andsecondarystructuredeterminationbyftir Introduction Proteinsareubiquitousinlivingorganismsandcells,andcanserveavarietyoffunctions.Proteinscanactas enzymes,hormones,antibiotics,receptors,orserveasstructuralsupportsintissuessuchasmuscle,hair,and
USP's Therapeutic Peptides Expert Panel discusses manufacturing processes and impurity control for synthetic peptide APIs.
 Control Strategies for Synthetic Therapeutic Peptide APIs Part III: Manufacturing Process Considerations By Brian Gregg,Aleksander Swietlow,Anita Y. Szajek,Harold Rode,Michael Verlander,Ivo Eggen USP's
Control Strategies for Synthetic Therapeutic Peptide APIs Part III: Manufacturing Process Considerations By Brian Gregg,Aleksander Swietlow,Anita Y. Szajek,Harold Rode,Michael Verlander,Ivo Eggen USP's
THE His Tag Antibody, mab, Mouse
 THE His Tag Antibody, mab, Mouse Cat. No. A00186 Technical Manual No. TM0243 Update date 01052011 I Description.... 1 II Key Features. 2 III Storage 2 IV Applications.... 2 V Examples - ELISA..... 2 VI
THE His Tag Antibody, mab, Mouse Cat. No. A00186 Technical Manual No. TM0243 Update date 01052011 I Description.... 1 II Key Features. 2 III Storage 2 IV Applications.... 2 V Examples - ELISA..... 2 VI
Structure Determination
 5 Structure Determination Most of the protein structures described and discussed in this book have been determined either by X-ray crystallography or by nuclear magnetic resonance (NMR) spectroscopy. Although
5 Structure Determination Most of the protein structures described and discussed in this book have been determined either by X-ray crystallography or by nuclear magnetic resonance (NMR) spectroscopy. Although
Combinatorial Chemistry and solid phase synthesis seminar and laboratory course
 Combinatorial Chemistry and solid phase synthesis seminar and laboratory course Topic 1: Principles of combinatorial chemistry 1. Introduction: Why Combinatorial Chemistry? Until recently, a common drug
Combinatorial Chemistry and solid phase synthesis seminar and laboratory course Topic 1: Principles of combinatorial chemistry 1. Introduction: Why Combinatorial Chemistry? Until recently, a common drug
Paper: 6 Chemistry 2.130 University I Chemistry: Models Page: 2 of 7. 4. Which of the following weak acids would make the best buffer at ph = 5.0?
 Paper: 6 Chemistry 2.130 University I Chemistry: Models Page: 2 of 7 4. Which of the following weak acids would make the best buffer at ph = 5.0? A) Acetic acid (Ka = 1.74 x 10-5 ) B) H 2 PO - 4 (Ka =
Paper: 6 Chemistry 2.130 University I Chemistry: Models Page: 2 of 7 4. Which of the following weak acids would make the best buffer at ph = 5.0? A) Acetic acid (Ka = 1.74 x 10-5 ) B) H 2 PO - 4 (Ka =
Application Note # LCMS-62 Walk-Up Ion Trap Mass Spectrometer System in a Multi-User Environment Using Compass OpenAccess Software
 Application Note # LCMS-62 Walk-Up Ion Trap Mass Spectrometer System in a Multi-User Environment Using Compass OpenAccess Software Abstract Presented here is a case study of a walk-up liquid chromatography
Application Note # LCMS-62 Walk-Up Ion Trap Mass Spectrometer System in a Multi-User Environment Using Compass OpenAccess Software Abstract Presented here is a case study of a walk-up liquid chromatography
Combinatorial Biochemistry and Phage Display
 Combinatorial Biochemistry and Phage Display Prof. Valery A. Petrenko Director - Valery Petrenko Instructors Galina Kouzmitcheva and I-Hsuan Chen Auburn 2006, Spring semester COMBINATORIAL BIOCHEMISTRY
Combinatorial Biochemistry and Phage Display Prof. Valery A. Petrenko Director - Valery Petrenko Instructors Galina Kouzmitcheva and I-Hsuan Chen Auburn 2006, Spring semester COMBINATORIAL BIOCHEMISTRY
In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates
 In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates Using the Explore Workflow on the AB SCIEX TripleTOF 5600 System A major challenge in proteomics
In-Depth Qualitative Analysis of Complex Proteomic Samples Using High Quality MS/MS at Fast Acquisition Rates Using the Explore Workflow on the AB SCIEX TripleTOF 5600 System A major challenge in proteomics
Guide to Reverse Phase SpinColumns Chromatography for Sample Prep
 Guide to Reverse Phase SpinColumns Chromatography for Sample Prep www.harvardapparatus.com Contents Introduction...2-3 Modes of Separation...4-6 Spin Column Efficiency...7-8 Fast Protein Analysis...9 Specifications...10
Guide to Reverse Phase SpinColumns Chromatography for Sample Prep www.harvardapparatus.com Contents Introduction...2-3 Modes of Separation...4-6 Spin Column Efficiency...7-8 Fast Protein Analysis...9 Specifications...10
WESTERN BLOTTING TIPS AND TROUBLESHOOTING GUIDE TROUBLESHOOTING GUIDE
 WESTERN BLOTTING TIPS AND TROUBLESHOOTING GUIDE TIPS FOR SUCCESSFUL WESTERB BLOTS TROUBLESHOOTING GUIDE 1. Suboptimal protein transfer. This is the most common complaint with western blotting and could
WESTERN BLOTTING TIPS AND TROUBLESHOOTING GUIDE TIPS FOR SUCCESSFUL WESTERB BLOTS TROUBLESHOOTING GUIDE 1. Suboptimal protein transfer. This is the most common complaint with western blotting and could
MULTIPLE CHOICE QUESTIONS
 MULTIPLE CHOICE QUESTIONS 1. Most components of energy conversion systems evolved very early; thus, the most fundamental aspects of energy metabolism tend to be: A. quite different among a diverse group
MULTIPLE CHOICE QUESTIONS 1. Most components of energy conversion systems evolved very early; thus, the most fundamental aspects of energy metabolism tend to be: A. quite different among a diverse group
Introduction to Proteomics
 Introduction to Proteomics Why Proteomics? Same Genome Different Proteome Black Swallowtail - larvae and butterfly Biological Complexity Yeast - a simple proteome 6,113 proteins = 344,855 tryptic peptides
Introduction to Proteomics Why Proteomics? Same Genome Different Proteome Black Swallowtail - larvae and butterfly Biological Complexity Yeast - a simple proteome 6,113 proteins = 344,855 tryptic peptides
2007 7.013 Problem Set 1 KEY
 2007 7.013 Problem Set 1 KEY Due before 5 PM on FRIDAY, February 16, 2007. Turn answers in to the box outside of 68-120. PLEASE WRITE YOUR ANSWERS ON THIS PRINTOUT. 1. Where in a eukaryotic cell do you
2007 7.013 Problem Set 1 KEY Due before 5 PM on FRIDAY, February 16, 2007. Turn answers in to the box outside of 68-120. PLEASE WRITE YOUR ANSWERS ON THIS PRINTOUT. 1. Where in a eukaryotic cell do you
Quaternary structure
 Quaternary structure Assembly of multiple polypeptide chains in one integral structure The arrangement of the subunits gives rise to a stable structure Subunits may be identical or different A common shorthand
Quaternary structure Assembly of multiple polypeptide chains in one integral structure The arrangement of the subunits gives rise to a stable structure Subunits may be identical or different A common shorthand
H H N - C - C 2 R. Three possible forms (not counting R group) depending on ph
 depending on ph") Amino acids - 0 common amino acids there are others found naturally but much less frequently - Common structure for amino acid - C, -N, and functional groups all attached to the alpha carbon N - C - C
Amino acids - 0 common amino acids there are others found naturally but much less frequently - Common structure for amino acid - C, -N, and functional groups all attached to the alpha carbon N - C - C
Peptide Bonds: Structure
 Peptide Bonds: Structure Peptide primary structure The amino acid sequence, from - to C-terminus, determines the primary structure of a peptide or protein. The amino acids are linked through amide or peptide
Peptide Bonds: Structure Peptide primary structure The amino acid sequence, from - to C-terminus, determines the primary structure of a peptide or protein. The amino acids are linked through amide or peptide
Technical Note. Roche Applied Science. No. LC 18/2004. Assay Formats for Use in Real-Time PCR
 Roche Applied Science Technical Note No. LC 18/2004 Purpose of this Note Assay Formats for Use in Real-Time PCR The LightCycler Instrument uses several detection channels to monitor the amplification of
Roche Applied Science Technical Note No. LC 18/2004 Purpose of this Note Assay Formats for Use in Real-Time PCR The LightCycler Instrument uses several detection channels to monitor the amplification of
1 The water molecule and hydrogen bonds in water
 The Physics and Chemistry of Water 1 The water molecule and hydrogen bonds in water Stoichiometric composition H 2 O the average lifetime of a molecule is 1 ms due to proton exchange (catalysed by acids
The Physics and Chemistry of Water 1 The water molecule and hydrogen bonds in water Stoichiometric composition H 2 O the average lifetime of a molecule is 1 ms due to proton exchange (catalysed by acids
Peptides: Synthesis and Biological Interest
 Peptides: Synthesis and Biological Interest Therapeutic Agents Therapeutic peptides approved by the FDA (2009-2011) 3 Proteins Biopolymers of α-amino acids. Amino acids are joined by peptide bond. They
Peptides: Synthesis and Biological Interest Therapeutic Agents Therapeutic peptides approved by the FDA (2009-2011) 3 Proteins Biopolymers of α-amino acids. Amino acids are joined by peptide bond. They
INSTRUCTION Probemaker
 INSTRUCTION Probemaker Instructions for Duolink In Situ Probemaker PLUS (Art. no. 92009-0020) and Duolink In Situ Probemaker MINUS (Art. no. 92010-0020) Table of content 1. Introduction 4 2. Applications
INSTRUCTION Probemaker Instructions for Duolink In Situ Probemaker PLUS (Art. no. 92009-0020) and Duolink In Situ Probemaker MINUS (Art. no. 92010-0020) Table of content 1. Introduction 4 2. Applications
Part A: Amino Acids and Peptides (Is the peptide IAG the same as the peptide GAI?)
") ChemActivity 46 Amino Acids, Polypeptides and Proteins 1 ChemActivity 46 Part A: Amino Acids and Peptides (Is the peptide IAG the same as the peptide GAI?) Model 1: The 20 Amino Acids at Biological p See
ChemActivity 46 Amino Acids, Polypeptides and Proteins 1 ChemActivity 46 Part A: Amino Acids and Peptides (Is the peptide IAG the same as the peptide GAI?) Model 1: The 20 Amino Acids at Biological p See
Some terms: An antigen is a molecule or pathogen capable of eliciting an immune response
 Overview of the immune system We continue our discussion of protein structure by considering the structure of antibodies. All organisms are continually subject to attack by microorganisms and viruses.
Overview of the immune system We continue our discussion of protein structure by considering the structure of antibodies. All organisms are continually subject to attack by microorganisms and viruses.
Interaktionen von RNAs und Proteinen
 Sonja Prohaska Computational EvoDevo Universitaet Leipzig June 9, 2015 Studying RNA-protein interactions Given: target protein known to bind to RNA problem: find binding partners and binding sites experimental
Sonja Prohaska Computational EvoDevo Universitaet Leipzig June 9, 2015 Studying RNA-protein interactions Given: target protein known to bind to RNA problem: find binding partners and binding sites experimental
Carbohydrates, proteins and lipids
 Carbohydrates, proteins and lipids Chapter 3 MACROMOLECULES Macromolecules: polymers with molecular weights >1,000 Functional groups THE FOUR MACROMOLECULES IN LIFE Molecules in living organisms: proteins,
Carbohydrates, proteins and lipids Chapter 3 MACROMOLECULES Macromolecules: polymers with molecular weights >1,000 Functional groups THE FOUR MACROMOLECULES IN LIFE Molecules in living organisms: proteins,
Aiping Lu. Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn
 Aiping Lu Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn Proteome and Proteomics PROTEin complement expressed by genome Marc Wilkins Electrophoresis. 1995. 16(7):1090-4. proteomics
Aiping Lu Key Laboratory of System Biology Chinese Academic Society APLV@sibs.ac.cn Proteome and Proteomics PROTEin complement expressed by genome Marc Wilkins Electrophoresis. 1995. 16(7):1090-4. proteomics
Chemistry 20 Chapters 15 Enzymes
 Chemistry 20 Chapters 15 Enzymes Enzymes: as a catalyst, an enzyme increases the rate of a reaction by changing the way a reaction takes place, but is itself not changed at the end of the reaction. An
Chemistry 20 Chapters 15 Enzymes Enzymes: as a catalyst, an enzyme increases the rate of a reaction by changing the way a reaction takes place, but is itself not changed at the end of the reaction. An
Lecture 8. Protein Trafficking/Targeting. Protein targeting is necessary for proteins that are destined to work outside the cytoplasm.
 Protein Trafficking/Targeting (8.1) Lecture 8 Protein Trafficking/Targeting Protein targeting is necessary for proteins that are destined to work outside the cytoplasm. Protein targeting is more complex
Protein Trafficking/Targeting (8.1) Lecture 8 Protein Trafficking/Targeting Protein targeting is necessary for proteins that are destined to work outside the cytoplasm. Protein targeting is more complex
CHAPTER 6 AN INTRODUCTION TO METABOLISM. Section B: Enzymes
 CHAPTER 6 AN INTRODUCTION TO METABOLISM Section B: Enzymes 1. Enzymes speed up metabolic reactions by lowering energy barriers 2. Enzymes are substrate specific 3. The active site in an enzyme s catalytic
CHAPTER 6 AN INTRODUCTION TO METABOLISM Section B: Enzymes 1. Enzymes speed up metabolic reactions by lowering energy barriers 2. Enzymes are substrate specific 3. The active site in an enzyme s catalytic
Myoglobin and Hemoglobin
 Myoglobin and Hemoglobin Myoglobin and hemoglobin are hemeproteins whose physiological importance is principally related to their ability to bind molecular oxygen. Myoglobin (Mb) The oxygen storage protein
Myoglobin and Hemoglobin Myoglobin and hemoglobin are hemeproteins whose physiological importance is principally related to their ability to bind molecular oxygen. Myoglobin (Mb) The oxygen storage protein
LC-MS/MS for Chromatographers
 LC-MS/MS for Chromatographers An introduction to the use of LC-MS/MS, with an emphasis on the analysis of drugs in biological matrices LC-MS/MS for Chromatographers An introduction to the use of LC-MS/MS,
LC-MS/MS for Chromatographers An introduction to the use of LC-MS/MS, with an emphasis on the analysis of drugs in biological matrices LC-MS/MS for Chromatographers An introduction to the use of LC-MS/MS,
Molecular Cell Biology. Prof. D. Karunagaran. Department of Biotechnology. Indian Institute of Technology Madras
 Molecular Cell Biology Prof. D. Karunagaran Department of Biotechnology Indian Institute of Technology Madras Module 5 Methods in Cell Biology (Methods to Manipulate Protein, DNA and RNA and Methods to
Molecular Cell Biology Prof. D. Karunagaran Department of Biotechnology Indian Institute of Technology Madras Module 5 Methods in Cell Biology (Methods to Manipulate Protein, DNA and RNA and Methods to
How many of you have checked out the web site on protein-dna interactions?
 How many of you have checked out the web site on protein-dna interactions? Example of an approximately 40,000 probe spotted oligo microarray with enlarged inset to show detail. Find and be ready to discuss
How many of you have checked out the web site on protein-dna interactions? Example of an approximately 40,000 probe spotted oligo microarray with enlarged inset to show detail. Find and be ready to discuss
Definition of the Measurand: CRP
 A Reference Measurement System for C-reactive Protein David M. Bunk, Ph.D. Chemical Science and Technology Laboratory National Institute of Standards and Technology Definition of the Measurand: Human C-reactive
A Reference Measurement System for C-reactive Protein David M. Bunk, Ph.D. Chemical Science and Technology Laboratory National Institute of Standards and Technology Definition of the Measurand: Human C-reactive
Amino Acids as Acids, Bases and Buffers:
 Amino Acids as Acids, Bases and Buffers: - Amino acids are weak acids - All have at least 2 titratable protons (shown below as fully protonated species) and therefore have 2 pka s o α-carboxyl (-COOH)
Amino Acids as Acids, Bases and Buffers: - Amino acids are weak acids - All have at least 2 titratable protons (shown below as fully protonated species) and therefore have 2 pka s o α-carboxyl (-COOH)
Transmembrane proteins span the bilayer. α-helix transmembrane domain. Multiple transmembrane helices in one polypeptide
 Transmembrane proteins span the bilayer α-helix transmembrane domain Hydrophobic R groups of a.a. interact with fatty acid chains Multiple transmembrane helices in one polypeptide Polar a.a. Hydrophilic
Transmembrane proteins span the bilayer α-helix transmembrane domain Hydrophobic R groups of a.a. interact with fatty acid chains Multiple transmembrane helices in one polypeptide Polar a.a. Hydrophilic
13C NMR Spectroscopy
 13 C NMR Spectroscopy Introduction Nuclear magnetic resonance spectroscopy (NMR) is the most powerful tool available for structural determination. A nucleus with an odd number of protons, an odd number
13 C NMR Spectroscopy Introduction Nuclear magnetic resonance spectroscopy (NMR) is the most powerful tool available for structural determination. A nucleus with an odd number of protons, an odd number
How To Understand Enzyme Kinetics
 Chapter 12 - Reaction Kinetics In the last chapter we looked at enzyme mechanisms. In this chapter we ll see how enzyme kinetics, i.e., the study of enzyme reaction rates, can be useful in learning more
Chapter 12 - Reaction Kinetics In the last chapter we looked at enzyme mechanisms. In this chapter we ll see how enzyme kinetics, i.e., the study of enzyme reaction rates, can be useful in learning more
SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays
 Protocol SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878 Order per fax: +49-30-6392-7888 or e-mail: www: peptide@jpt.com www.jpt.com
Protocol SpikeTides TM Peptides for relative and absolute quantification in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878 Order per fax: +49-30-6392-7888 or e-mail: www: peptide@jpt.com www.jpt.com
Chapter 6: Antigen-Antibody Interactions
 Chapter 6: Antigen-Antibody Interactions I. Strength of Ag-Ab interactions A. Antibody Affinity - strength of total noncovalent interactions between single Ag-binding site on an Ab and a single epitope
Chapter 6: Antigen-Antibody Interactions I. Strength of Ag-Ab interactions A. Antibody Affinity - strength of total noncovalent interactions between single Ag-binding site on an Ab and a single epitope
1. A covalent bond between two atoms represents what kind of energy? a. Kinetic energy b. Potential energy c. Mechanical energy d.
 1. A covalent bond between two atoms represents what kind of energy? a. Kinetic energy b. Potential energy c. Mechanical energy d. Solar energy A. Answer a is incorrect. Kinetic energy is the energy of
1. A covalent bond between two atoms represents what kind of energy? a. Kinetic energy b. Potential energy c. Mechanical energy d. Solar energy A. Answer a is incorrect. Kinetic energy is the energy of
Optimal Conditions for F(ab ) 2 Antibody Fragment Production from Mouse IgG2a
 2 Antibody Fragment Production from Mouse IgG2a") Optimal Conditions for F(ab ) 2 Antibody Fragment Production from Mouse IgG2a Ryan S. Stowers, 1 Jacqueline A. Callihan, 2 James D. Bryers 2 1 Department of Bioengineering, Clemson University, Clemson,
Optimal Conditions for F(ab ) 2 Antibody Fragment Production from Mouse IgG2a Ryan S. Stowers, 1 Jacqueline A. Callihan, 2 James D. Bryers 2 1 Department of Bioengineering, Clemson University, Clemson,
CHAPTER 4: Enzyme Structure ENZYMES
 CHAPTER 4: ENZYMES Enzymes are biological catalysts. There are about 40,000 different enzymes in human cells, each controlling a different chemical reaction. They increase the rate of reactions by a factor
CHAPTER 4: ENZYMES Enzymes are biological catalysts. There are about 40,000 different enzymes in human cells, each controlling a different chemical reaction. They increase the rate of reactions by a factor
Protein Melting Curves
 Protein Melting Curves In previous classes we talked a lot about what aspects of a protein structure stabilize a protein and what aspects destabilize it. But how would one actually test such predictions?
Protein Melting Curves In previous classes we talked a lot about what aspects of a protein structure stabilize a protein and what aspects destabilize it. But how would one actually test such predictions?
Lecture 15: Enzymes & Kinetics Mechanisms
 ROLE OF THE TRANSITION STATE Lecture 15: Enzymes & Kinetics Mechanisms Consider the reaction: H-O-H + Cl - H-O δ- H Cl δ- HO - + H-Cl Reactants Transition state Products Margaret A. Daugherty Fall 2004
ROLE OF THE TRANSITION STATE Lecture 15: Enzymes & Kinetics Mechanisms Consider the reaction: H-O-H + Cl - H-O δ- H Cl δ- HO - + H-Cl Reactants Transition state Products Margaret A. Daugherty Fall 2004
green B 1 ) into a single unit to model the substrate in this reaction. enzyme
 into a single unit to model the substrate in this reaction. enzyme") Teacher Key Objectives You will use the model pieces in the kit to: Simulate enzymatic actions. Explain enzymatic specificity. Investigate two types of enzyme inhibitors used in regulating enzymatic activity.
Teacher Key Objectives You will use the model pieces in the kit to: Simulate enzymatic actions. Explain enzymatic specificity. Investigate two types of enzyme inhibitors used in regulating enzymatic activity.
Antibody Function & Structure
 Antibody Function & Structure Specifically bind to antigens in both the recognition phase (cellular receptors) and during the effector phase (synthesis and secretion) of humoral immunity Serology: the
Antibody Function & Structure Specifically bind to antigens in both the recognition phase (cellular receptors) and during the effector phase (synthesis and secretion) of humoral immunity Serology: the
The peptide bond is rigid and planar
 Level Description Bonds Primary Sequence of amino acids in proteins Covalent (peptide bonds) Secondary Structural motifs in proteins: α- helix and β-sheet Hydrogen bonds (between NH and CO groups in backbone)
Level Description Bonds Primary Sequence of amino acids in proteins Covalent (peptide bonds) Secondary Structural motifs in proteins: α- helix and β-sheet Hydrogen bonds (between NH and CO groups in backbone)
Enzymes: Practice Questions #1
 Enzymes: Practice Questions #1 1. Compound X increases the rate of the reaction below. Compound X is most likely A. an enzyme B. a lipid molecule C. an indicator D. an ADP molecule 2. The equation below
Enzymes: Practice Questions #1 1. Compound X increases the rate of the reaction below. Compound X is most likely A. an enzyme B. a lipid molecule C. an indicator D. an ADP molecule 2. The equation below
Table of contents. Bibliografische Informationen http://d-nb.info/1006571213. digitalisiert durch
 1. Research scope: The role of structure rigid'tf'ication in nature and chemistry 1 2. Establishing a Dha=Tap backbone scan in order to elucidate structural properties of the N-terminusofNPY 5 2.1 Introduction.
1. Research scope: The role of structure rigid'tf'ication in nature and chemistry 1 2. Establishing a Dha=Tap backbone scan in order to elucidate structural properties of the N-terminusofNPY 5 2.1 Introduction.
How To Make A Drug From A Peptide
 MODERN PERSPECTIVES ON PEPTIDE SYNTHESIS INTRODUCTION WHITEPAPER www.almacgroup.com The complexity of synthetic peptide products, whether as reagents used in research or as therapeutic APIs, is increasing.
MODERN PERSPECTIVES ON PEPTIDE SYNTHESIS INTRODUCTION WHITEPAPER www.almacgroup.com The complexity of synthetic peptide products, whether as reagents used in research or as therapeutic APIs, is increasing.
Copyright 2000-2003 Mark Brandt, Ph.D. 54
 Pyruvate Oxidation Overview of pyruvate metabolism Pyruvate can be produced in a variety of ways. It is an end product of glycolysis, and can be derived from lactate taken up from the environment (or,
Pyruvate Oxidation Overview of pyruvate metabolism Pyruvate can be produced in a variety of ways. It is an end product of glycolysis, and can be derived from lactate taken up from the environment (or,
Topic 2: Energy in Biological Systems
 Topic 2: Energy in Biological Systems Outline: Types of energy inside cells Heat & Free Energy Energy and Equilibrium An Introduction to Entropy Types of energy in cells and the cost to build the parts
Topic 2: Energy in Biological Systems Outline: Types of energy inside cells Heat & Free Energy Energy and Equilibrium An Introduction to Entropy Types of energy in cells and the cost to build the parts
Nafith Abu Tarboush DDS, MSc, PhD natarboush@ju.edu.jo www.facebook.com/natarboush
 Nafith Abu Tarboush DDS, MSc, PhD natarboush@ju.edu.jo www.facebook.com/natarboush α-keratins, bundles of α- helices Contain polypeptide chains organized approximately parallel along a single axis: Consist
Nafith Abu Tarboush DDS, MSc, PhD natarboush@ju.edu.jo www.facebook.com/natarboush α-keratins, bundles of α- helices Contain polypeptide chains organized approximately parallel along a single axis: Consist
Determination of Equilibrium Constants using NMR Spectrscopy
 CHEM 331L Physical Chemistry Laboratory Revision 1.0 Determination of Equilibrium Constants using NMR Spectrscopy In this laboratory exercise we will measure a chemical equilibrium constant using key proton
CHEM 331L Physical Chemistry Laboratory Revision 1.0 Determination of Equilibrium Constants using NMR Spectrscopy In this laboratory exercise we will measure a chemical equilibrium constant using key proton
Refinement of a pdb-structure and Convert
 Refinement of a pdb-structure and Convert A. Search for a pdb with the closest sequence to your protein of interest. B. Choose the most suitable entry (or several entries). C. Convert and resolve errors
Refinement of a pdb-structure and Convert A. Search for a pdb with the closest sequence to your protein of interest. B. Choose the most suitable entry (or several entries). C. Convert and resolve errors